
About this Transcript 🔗
This document is a transcript of an official FDA (or IMDRF) guidance document. We transcribe the official PDFs into HTML so that we can share links to particular sections of the guidance when communicating internally and with our clients. We do our best to be accurate and have a thorough review process, but occasionally mistakes slip through. If you notice a typo, please email a screenshot of it to Mihajlo at mgrcic@innolitics.com so we can fix it.
Preamble 🔗
Document issued on July 28, 2014.
For questions for the Center for Devices and Radiological Health regarding this document, contact the Premarket Notification (510(k)) Section at 301-796-5640.
For questions for the Center for Biologics Evaluation and Research regarding this document, contact the Office of Communication, Outreach and Development at 1-800-335-4709 or 240-402-7800.
I. Introduction 🔗
FDA developed this document to provide guidance to industry and FDA staff about current review practices for premarket notification (510(k)) submissions. The intent of this guidance is to identify, explain, and clarify each of the critical decision points in the decision-making process FDA uses to determine substantial equivalence. This guidance is not intended to implement significant policy changes to the current 510(k) review process. Rather, the intent of this guidance is to enhance the predictability, consistency, and transparency of the 510(k) program by describing in greater detail the regulatory framework, policies, and practices underlying FDA’s 510(k) review.
The draft of this guidance document contained sections addressing FDA’s Special and Abbreviated 510(k) programs. FDA intends to finalize those sections separately. Until FDA issues new final recommendations on the Special and Abbreviated 510(k) programs, the recommendations for Special and Abbreviated 510(k)s contained in “The New 510(k) Paradigm - Alternate Approaches to Demonstrating Substantial Equivalence in Premarket Notifications,” dated March 20, 1998, (http://www.fda.gov/MedicalDevices/DeviceRegulationandGuidance/GuidanceDocuments/ucm080187.htm) remain in effect.
FDA's guidance documents, including this guidance, do not establish legally enforceable responsibilities. Instead, guidances describe the Agency's current thinking on a topic and should be viewed only as recommendations, unless specific regulatory or statutory requirements are cited. The use of the word should in Agency guidances means that something is suggested or recommended, but not required.
II. Background 🔗
A. The Medical Device Amendments and Device Classification 🔗
The Medical Device Amendments (MDA) (Pub. L. 94-295) to the Federal Food, Drug, and Cosmetic (FD&C) Act were enacted on May 28, 1976. The MDA directed FDA to issue regulations that classify all devices that were in commercial distribution at that time into one of three regulatory control categories: Class I, II, or III, depending upon the degree of regulation necessary to provide reasonable assurance of their safety and effectiveness. The class into which a device is placed determines the requirements that a medical device manufacturer must meet prior to distributing a device in interstate commerce. According to section 513(a)(1) of the FD&C Act (21 U.S.C. § 360c(a)(1)), the three device classes are defined as follows:
- Class I: Devices are subject to a comprehensive set of regulatory authorities called general controls that are applicable to all classes of devices.1
- Class II: Devices for which general controls, by themselves, are insufficient to provide reasonable assurance of the safety and effectiveness of the device, and for which there is sufficient information to establish special controls to provide such assurance.2
- Class III: Devices for which general controls, by themselves, are insufficient and for which there is insufficient information to establish special controls to provide reasonable assurance of the safety and effectiveness of the device. Class III devices typically require premarket approval.3
Premarket notification is the process by which a new device,4 i.e., a post-amendments device, is classified into one of these three device classes.5 A manufacturer who intends to market in the United States a Class I, II, or III device intended for human use, for which a Premarket Approval application (PMA) is not required, must submit to FDA a premarket notification submission (often referred to as a 510(k)), unless the device is exempt from the 510(k) requirements of the FD&C Act and does not exceed the limitations of exemptions for each of the device classification regulations (Section 9 of 21 CFR Parts 862 through 892, e.g., 21 CFR 862.9, 21 CFR 864.9, etc.). Under section 510(k) of the FD&C Act, a manufacturer must submit a 510(k) to FDA at least 90 days before introducing, or delivering for introduction, a device into interstate commerce for commercial distribution so the Agency can determine whether or not the device meets the criteria for market clearance (Sections 510(k) and (n) of the FD&C Act (21 U.S.C. §§ 360(k) & (n))). The Agency bases its decision on whether the device is substantially equivalent (SE) to a legally marketed (predicate) device (Section 513(i) of the FD&C Act (21 U.S.C. § 360c(i))). The device cannot be commercialized until FDA issues an order (510(k) clearance) stating that the device has been determined to be SE (Section 513(f)(1) of the FD&C Act (21 U.S.C. § 360c(f)(1))).
B. The 510(k) Classification Process 🔗
According to section 513(f) of the FD&C Act, a new (i.e., post-amendments) device is automatically in Class III and must undergo premarket approval or reclassification before it can be marketed, unless it is a type of device that was in commercial distribution prior to May 28, 1976, and is SE to another such device; or it is within a type of device introduced after May 28, 1976, that has been reclassified into Class I or II and is SE to another device within such classification. For information about how FDA’s classification product codes assist in accurate identification and tracking of current medical devices, please see FDA’s Guidance for Industry and Food and Drug Administration Staff, “Medical Device Classification Product Codes” (http://www.fda.gov/MedicalDevices/DeviceRegulationandGuidance/GuidanceDocuments/ucm2853 17.htm).
When FDA determines under sections 510(k), 513(f)(1), and 513(i) of the FD&C Act that a new device is SE to a legally marketed (predicate) device, the new device is classified into the same class and subject to the same requirements as the predicate device. (See Section IV.C.) A determination that a new device is not substantially equivalent (NSE) to a predicate device results in the new device being classified into Class III. Thus, 510(k) review is both the mechanism by which a manufacturer seeks marketing authorization for a new device and by which FDA classifies devices into their appropriate regulatory category. Because devices are classified according to the level of regulatory control necessary to provide a reasonable assurance of safety and effectiveness,6 classification of a new device through the 510(k) process requires FDA to determine the issues of safety and effectiveness presented by the new device, and the regulatory controls necessary to address those issues.7
C. Evolution of the 510(k) Program 🔗
Since its inception, the 510(k) program has undergone a number of statutory changes. Notably, the Safe Medical Devices Act of 1990 (Pub. L. 101-629) added section 513(i), which codified FDA review practice in applying the “substantial equivalence” review standard. In addition, FDA has modified its implementation of the program to adapt to changing circumstances and to accommodate the evolving medical device landscape. For example, the alternative options of a Special 510(k) or an Abbreviated 510(k) still exist today. Additional information regarding these alternative options can be found in FDA’s guidance, “The New 510(k) Paradigm – Alternative Approaches to Demonstrating Substantial Equivalence in Premarket Notifications” (http://www.fda.gov/downloads/MedicalDevices/DeviceRegulationandGuidance/GuidanceDocuments/ucm080189.pdf). The current 510(k) program reflects the current statutory framework and FDA’s implementation of that framework through regulation, guidance, and administrative practice. A history of the 510(k) program has been summarized in other documents that FDA has published.8
This guidance document provides updated information to the existing guidance document entitled “Guidance on the CDRH Premarket Notification Review Program, 510(k) Memorandum K86-3” (K86-3 Guidance), issued on June 30, 1986. The K86-3 Guidance was written and issued as final guidance prior to the February 27, 1997 implementation of FDA’s Good Guidance Practices (GGPs), and has not been updated since its initial publication date. This guidance replaces the K86-3 Guidance.
III. Scope 🔗
This guidance provides recommendations to industry and FDA staff about the content of 510(k) submissions and the decision-making process for determining substantial equivalence of devices reviewed under the 510(k) program. The guidance has been organized to coincide with the critical decision points outlined in the 510(k) Decision-Making Flowchart (See Appendix A), which has been updated to track section 513(i) of the FD&C Act and relevant regulations more closely. This document provides guidance on the following issues:
- the appropriate use of multiple predicates (See Section IV.C);
- the processes associated with determining whether a new device with new indications for use has a new intended use (See Section IV.D);
- the process for determining whether different technological characteristics raise different questions of safety and effectiveness (See Section IV.E);
- when performance data, with special emphasis on clinical performance data, may be necessary to support an SE determination (See Section IV.F); and
- how to develop 510(k) Summaries to promote greater transparency in the 510(k) decision-making process (See Section IV.G).
The overarching principles in this guidance are applicable to devices that are subject to 510(k) review by CDRH, including the Office of Device Evaluation (ODE) and the Office of In Vitro Diagnostics and Radiological Health (OIR), as well as devices that are subject to 510(k) review by the Center for Biologics Evaluation and Research (CBER). This guidance is not intended to supplant existing device-specific guidance, but may cover broader areas not addressed in device-specific guidance documents. If you have questions about how this guidance and a device-specific guidance apply to a particular issue, please contact FDA to discuss. In addition, this guidance does not address review issues unique to combination products. For information on combination products, please refer to the Office of Combination Products webpage (http://www.fda.gov/AboutFDA/CentersOffices/OfficeofMedicalProductsandTobacco/OfficeofScien ceandHealthCoordination/ucm2018184.htm).
IV. The 510(k) Decision-Making Process 🔗
A 510(k) is a premarket submission made to FDA to demonstrate that the new device to be marketed is “substantially equivalent” to a legally marketed device9 (21 U.S.C. §§ 360(k), 360(n), 360c(f)(1) & 360c(i); 21 CFR 807.92(a)(3)) which is not subject to PMA. Manufacturers must compare their new device to a similar legally marketed device to support its substantial equivalence (21 U.S.C. § 360c(i); 21 CFR 807.92(a)(3)).
The most commonly used method of demonstrating substantial equivalence is through the submission and FDA review and clearance of a Traditional 510(k). Under 21 CFR 807.87, FDA established basic content requirements for 510(k)s to be submitted by device manufacturers in support of substantial equivalence. The Agency has provided a general framework on how to format an original submission for a Traditional 510(k) in FDA’s Guidance for Industry and FDA Staff, “Format for Traditional and Abbreviated 510(k)s” (http://www.fda.gov/MedicalDevices/DeviceRegulationandGuidance/GuidanceDocuments/ucm0843 65.htm). Although the basic content requirements apply to all 510(k)s, the type of data and information necessary to establish substantial equivalence varies by the type of device and the differences between the new device and the predicate device. FDA has issued many device-specific guidance documents that clarify the data that should be included in 510(k)s for particular device types. If a manufacturer is unsure of what information to include within a 510(k) submission, the manufacturer may contact FDA and submit a pre-submission to seek additional feedback to ensure submissions contain appropriate data elements. For more information on the pre-submission process, see FDA’s Guidance for Industry and FDA Staff, “Requests for Feedback on Medical Device Submissions: The Pre-Submission Program and Meetings with Food and Drug Administration Staff” (http://www.fda.gov/downloads/MedicalDevices/DeviceRegulationandGuidance/GuidanceDocuments/UCM311176.pdf).
Please note that the use of the Standards Data Report for 510(k)s (Form 3654) (http://www.fda.gov/downloads/AboutFDA/ReportsManualsForms/Forms/UCM081667.pdf), recognized consensus standards, and device-specific guidance documents is not limited to Abbreviated 510(k) submissions. Appropriate reliance on these documents can facilitate the review of all 510(k) submissions and can help to make the review process more consistent. Medical device manufacturers should consider relying on and citing to standards and device-specific guidance documents wherever appropriate, regardless of the type of 510(k) submission.
A new device does not need to be identical to the predicate device for it to be found substantially equivalent to the predicate device. In FDA’s experience, it is rare for a new device to be identical to a predicate device. Given the diversity of technologies evaluated under this review standard, this guidance adopts a flexible approach to determining “substantial equivalence” to accommodate evolving technology while maintaining predictability and consistency to promote confidence among device developers, practitioners, and patients.
A. The 510(k) Review Standard 🔗
-
The Statutory Standard The 510(k) review standard (substantial equivalence of a new device to a legally marketed (predicate) device) differs from the PMA review standard (reasonable assurance of safety and effectiveness). The 510(k) review standard is comparative, whereas the PMA standard relies on an independent demonstration of safety and effectiveness. Nonetheless, the principles of safety and effectiveness underlie the substantial equivalence determination in every 510(k) review. The standard for a determination of substantial equivalence in a 510(k) review is set out in section 513(i) of the FD&C Act, which states:
Substantial Equivalence
(i)(1)(A) For purposes of determinations of substantial equivalence under subsection (f) and section 520(l), the term "substantially equivalent" or "substantial equivalence" means, with respect to a device being compared to a predicate device, that the device has the same intended use as the predicate device and that the Secretary by order has found that the device (i) has the same technological characteristics as the predicate device, or
(ii)(I) has different technological characteristics and the information submitted that the device is substantially equivalent to the predicate device contains information, including appropriate clinical or scientific data if deemed necessary by the Secretary or a person accredited under section 523, that demonstrates that the device is as safe and effective as a legally marketed device, and (II) does not raise different questions of safety and effectiveness than the predicate device.
(B) For purposes of subparagraph (A), the term “different technological characteristics” means, with respect to a device being compared to a predicate device, that there is a significant change in the materials, design, energy source, or other features of the device from those of the predicate device.
Safety and effectiveness factor into both parts of the FDA’s review. First, FDA must find that the intended use of the device and its predicate are “the same.” As discussed in the Intended Use Section of this guidance, differences in the indications for use, such as the population for which a device is intended or the disease a device is intended to treat do not necessarily result in a new intended use. Such differences result in a new intended use when they affect (or may affect) the safety and/or effectiveness of the new device as compared to the predicate device and the differences cannot be adequately evaluated under the comparative standard of substantial equivalence. (See Section IV.D.)
Second, when comparing a new device to a predicate device, FDA must find that the two devices have “the same technological characteristics,” or that a “significant change in the materials, design, energy source or other features of the device” does not raise different questions of safety and effectiveness and that the device is as safe and effective as a legally marketed device.
Although the 510(k) process involves a comparison of a new device to a predicate device rather than an independent demonstration of the new device’s safety and effectiveness, as is required for approval of a PMA, in both cases FDA’s review decision reflects a determination of the level of control necessary to provide a “reasonable assurance of safety and effectiveness.”10 The evidentiary standard, however, is different. In the 510(k) context, FDA generally relies, in part, on FDA’s prior determination that a reasonable assurance of safety and effectiveness exists for the predicate device. Demonstrating basic similarities between a new device and a predicate device typically requires manufacturers to provide descriptive information such as a comparison of specifications, materials, and technology. In contrast, FDA generally evaluates differences between the new device and the predicate device to determine their effect on safety and effectiveness. It follows that the evidence necessary to show substantial equivalence will increase as differences between the new device and the predicate device increase if those differences significantly affect, or may significantly affect, safety or effectiveness (21 CFR 807.81).
-
The Least Burdensome Principle The FDA Modernization Act of 1997 (FDAMA) added two provisions, commonly known as “the least burdensome provisions,” to the FD&C Act; these were amended by the FDA Safety and Innovation Act of 2012 (FDASIA) (Pub. L. 112-144; 126 Stat. 1051). The provision relating to substantial equivalence, section 513(i)(1)(D), states:
- Whenever the Secretary requests information to demonstrate that devices with differing technological characteristics are substantially equivalent, the Secretary shall only request information that is necessary to making substantial equivalence determinations. In making such request, the Secretary shall consider the least burdensome means of demonstrating substantial equivalence and request information accordingly.
- For purposes of clause (i), the term “necessary” means the minimum required information that would support a determination of substantial equivalence between a new device and a predicate device.
- Nothing in this subparagraph shall alter the standard for determining substantial equivalence between a new device and a predicate device.
Although the statutory provision refers only to information requests related to determining the substantial equivalence of technological characteristics of a device and its predicate, the underlying principle that information requests should relate to the review standard is a basic principle of good regulatory practice with broad applicability to the 510(k) decision-making process.
FDA’s guidances, “The Least Burdensome Provisions of the FDA Modernization Act of 1997: Concept and Principles” (http://www.fda.gov/MedicalDevices/DeviceRegulationandGuidance/GuidanceDocuments/ucm0859 94.htm) and “Suggested Format for Developing and Responding to Deficiencies in Accordance with the Least Burdensome Provisions of FDAMA” (http://www.fda.gov/MedicalDevices/DeviceRegulationandGuidance/GuidanceDocuments/ucm0736 79.htm) (“the Least Burdensome Guidances”), explain how FDA intends to apply the least burdensome provisions. “The Least Burdensome Provisions of the FDA Modernization Act of 1997: Concept and Principles” interprets least burdensome as “a successful means of addressing a premarket issue that involves the most appropriate investment of time, effort, and resources on the part of industry and FDA,” and specifies that the least burdensome provisions do not affect the statutory premarket review standards for devices. The recommendations discussed in this guidance for evaluating substantial equivalence are consistent with the principles discussed in the Least Burdensome Guidances, but applies them by discussing the considerations that may affect the type of information necessary to demonstrate substantial equivalence at different decision points in the review of a 510(k).
-
Categories of NSE Determinations The K86-3 Guidance stated: “If it is clear from an initial review that a new device has a[n] intended use or technological feature that makes it NSE, the Center will not review or require performance information in the 510(k). Instead the applicant will be notified that the device is NSE, and any performance data will be reviewed in a PMA or reclassification petition.” The same is not true for NSE decisions based on a lack of performance data, which do not preclude submission of a new 510(k) containing different or additional data to support a finding of substantial equivalence. Thus, it has been FDA’s longstanding policy to treat NSE determinations as falling into two categories: (1) those that reflect FDA’s affirmative determination that the device is a Class III device and cannot be reviewed in a 510(k) submission, and (2) those that reflect inadequacies in the evidence that preclude a finding of substantial equivalence.
The first category of NSE determinations includes a variety of different decisions, such as a finding of a lack of a predicate device, a new intended use, or different technological characteristics that raise different questions of safety or effectiveness when the new device is compared to the cited predicate device, that as a matter of law results in an NSE determination. In most cases, FDA will provide the opportunity for the manufacturer to respond to initial concerns regarding the equivalency of the new device’s intended use or technology to a predicate device via response to a request for additional information. When FDA issues an NSE letter for a reason in this first category, the letter will typically not identify performance-based deficiencies. Consequently, the device is automatically classified into Class III and will require PMA approval,11 or if eligible, granting of a De Novo before marketing. If FDA believes that the device found NSE may be eligible for the De Novo program, the NSE letter will typically indicate FDA’s recommendation. More information regarding the De Novo program can be found in FDA’s Guidance for Industry and CDRH Staff, “New Section 513(f)(2) - Evaluation of Automatic Class III Designation” (http://www.fda.gov/downloads/MedicalDevices/DeviceRegulationandGuidance/GuidanceDocument s/ucm080197.pdf).
The second category of NSE determinations is for those devices for which FDA has not affirmed that the new device has a different intended use or that the different technological characteristics raise different questions of safety or effectiveness when compared to the cited predicate device, but rather that the information provided in the submission is insufficient to demonstrate substantial equivalence to the predicate device. In this situation, FDA generally first identifies the specific additional information – typically related to performance testing – that needs to be provided so that FDA may complete its evaluation of substantial equivalence. Upon receipt of FDA’s request for additional information (either through a formal letter, email, phone call or fax), the manufacturer has the opportunity to respond to FDA’s request. If the manufacturer in its response does not provide the requested information or a substantive justification for not providing the requested information, FDA will consider the response incomplete and place the submission immediately back on hold as an incomplete response. Once a complete response is received, FDA will work with the manufacturer to try to resolve identified deficiencies in an interactive capacity following the timeframes and interactions instituted with the passage of the Medical Device User Fee Amendments of 2012 (MDUFA III).12 For more information on communications during the review of a 510(k) submission, see FDA’s Guidance for Industry and FDA Staff, “Types of Communication During the Review of Medical Device Submissions” (http://www.fda.gov/MedicalDevices/DeviceRegulationandGuidance/GuidanceDocuments/ucm341918.htm). If the manufacturer does not respond at all to FDA’s requests for additional information, the submission will be subsequently withdrawn by FDA within the timeframe specified by FDA’s Guidance for Industry and FDA Staff, “FDA and Industry Actions on Premarket Notification (510(k)) Submissions: Effect on FDA Review Clock and Goals” (http://www.fda.gov/downloads/MedicalDevices/DeviceRegulationandGuidance/GuidanceDocuments/UCM089738.pdf). If a 510(k) is withdrawn due to a lack of response, the manufacturer may submit a new 510(k) with additional information that addresses the outstanding deficiencies communicated by FDA based on the review of the prior 510(k). If the manufacturer provides the requested information after the withdrawal date, it will be considered and processed as a new 510(k) (21 CFR 807.87(l)); therefore, all information previously submitted would have to be resubmitted so that the new 510(k) is complete. If a new 510(k) is submitted to address deficiencies raised in this type of NSE letter, as explained in FDA’s Guidance for Industry and FDA Staff, “Refuse to Accept Policy for 510(k)s” (http://www.fda.gov/ucm/groups/fdagov-public/@fdagov-meddevgen/documents/document/ucm315014.pdf), the new 510(k) should clearly identify how the outstanding issues have been addressed and cross-reference where the new information is provided within the newly submitted 510(k). Failure to cite the prior 510(k) number may result in a Refuse to Accept decision.
B. The Flowchart 🔗
The 510(k) Substantial Equivalence Decision-Making Process Flowchart (K86-3 Flowchart) was originally presented in the K86-3 Guidance and has served as the overarching “framework” for 510(k) decision-making for decades. The K86-3 Flowchart has provided a concise summary of the 510(k) decision-making process and serves as a common frame of reference for scientific and regulatory discussions related to the 510(k) process. However, the K86-3 Flowchart has not been updated since 1986 and, consequently, does not incorporate certain terminology set out in subsequent amendments to the FD&C Act. Furthermore, the K86-3 Flowchart’s visual structure may be more complex than necessary. To specifically address these issues, a modified Flowchart is provided that both more closely tracks the language of section 513(i) of the FD&C Act and relevant regulations, and visually simplifies our presentation of the decision-making algorithm.
It should be noted that the 510(k) Decision-Making Flowchart (the Flowchart) (see Appendix A) is meant to be used in conjunction with this guidance document and not as a “stand-alone” document without appropriate references to the context of each critical decision point.
C. Predicate Device(s) 🔗
As discussed in Section IV.A, the 510(k) review standard is substantial equivalence of a new device to a legally marketed device. Under 21 CFR 807.92(a)(3), a legally marketed device is a device that (i) was legally marketed prior to May 28, 1976 (preamendments device13 ) and for which a PMA is not required; or (ii) has been reclassified from Class III to Class II or I; or (iii) has been found SE through the 510(k) process. For purposes of determining substantial equivalence, the legally marketed device is commonly referred to as the “predicate device” or “predicate.” While manufacturers may identify more than one predicate device, only one is required. FDA encourages manufacturers to identify a single predicate device to simplify and facilitate the decision-making process. When a manufacturer does identify multiple predicates, the primary predicate refers to the one with indications for use and technological characteristics most similar to the device under review. Although using a single predicate is optimal, when multiple predicates are appropriate (as described in the examples below), FDA recommends identifying a primary predicate in the submission to facilitate a timely, well-supported decision.
Section 513(i) of the FD&C Act and 21 CFR 807.100(b) state that, for a new device to be considered substantially equivalent to a predicate device, the new device must have the same intended use as the predicate device and the same technological characteristics or different technological characteristics that do not raise different questions of safety and effectiveness than the predicate device. Therefore, the use of a “split predicate” is inconsistent with the 510(k) regulatory standard. “Split predicate” refers to a situation in which a manufacturer is attempting to “split” the 510(k) decision making process by demonstrating that a new device has the same intended use as one marketed device while comparing the new device’s technological characteristics with a second marketed device that has a different intended use. As a general matter, to find a device substantially equivalent, FDA must be able to address Decision Points 1 through 4 in the Flowchart using one predicate device identified by the manufacturer. FDA may use one or more additional devices proposed by the manufacturer in certain instances to help support substantial equivalence, as described below.
-
Multiple Predicates A manufacturer may use multiple predicate devices14 to help demonstrate substantial equivalence in certain circumstances. Manufacturers sometimes choose to do this when combining features from two or more predicate devices with the same intended use into a single new device, when seeking to market a device with more than one intended use, or when seeking more than one indication for use under the same intended use, as described in the examples below.
Multiple Predicates Example 1:
A manufacturer submits a 510(k) for a new hemodialysis catheter. This new catheter has an extension (the portion of the device outside the body) design that is similar to predicate A and a tip (the portion of the device inside the body) design similar to predicate B. Both predicates A and B have the same intended use as the new device. In this example, the manufacturer is relying on both predicate A and predicate B, which have the same intended use as the new device, to support substantial equivalence with respect to technological characteristics. The manufacturer may choose either predicate as the primary predicate in this example.
Multiple Predicates Example 2:
A manufacturer submits a 510(k) for a plate indicated for fixation of both diaphyseal (the shaft of a long bone) and epiphyseal (the ends of a long bone) fractures, i.e., the plate can be used to set a long bone, such as the femur or thigh bone, that is broken in the middle or at the ends. The manufacturer cites a predicate device that is a plate indicated for middle bone fractures only and another predicate device that is indicated specifically for bone tip fractures. While the indications for use of each predicate device are different, both devices have the same intended use, namely, fracture fixation of the long bone.15 Thus, although the manufacturer could have used a single predicate device, in cases where a manufacturer intends to market a device for more than one indication and a different predicate exists to support each specific indication, the manufacturer may cite more than one relevant predicate device to support an SE determination. In this case, using two appropriate predicates clearly identified by the manufacturer helped to facilitate clearance of the new device, which was indicated to treat both types of fractures treated by the predicates.
Multiple Predicates Example 3:
A manufacturer submits a 510(k) for a laser platform that consists of two hand pieces: an Er:YAG laser hand piece and a Q-Switch Nd:YAG laser hand piece. The manufacturer cites two predicates to support substantial equivalence for both of their requested proposed indications for use. In this case, each predicate cited does not share the same indications for use as the other predicate because each predicate consists of only one hand piece in which the indications correspond to the indications for one of the hand pieces included in the new device. However, the indications for both hand pieces fall within the scope of the general intended use of lasers, “incision, excision, ablation, vaporization of soft tissue.” The Er:YAG laser hand piece is indicated for the incision, excision, ablation, vaporization of soft tissue; and the Q-Switch Nd:YAG laser hand piece is indicated for tattoo removal. The new device is found substantially equivalent to the predicate devices because it has the same intended use and the new device’s technological characteristics are similar to the cited predicates.
In each example above, a single predicate could have been used to establish substantial equivalence of the new device, but the manufacturer used multiple predicates to show that FDA had found similar technology or indications to be substantially equivalent.
Multiple Predicates Example 4:
A manufacturer submits a 510(k) for a multi-parameter monitor. The monitor includes different technologies that can stand alone independently, but can also be used together for the general intended use of measuring patient vital information. If there is a predicate device for each of the parameters, then the combination of these parameters, assuming that monitoring of each individual parameter does not interfere with the others, can be found substantially equivalent.
It should be noted that in Examples 2, 3, and 4 above, the specific indications of the new device may necessitate new performance testing, but they do not change the overall intended use of the device relative to the predicates. These types of situations will need to be assessed on a case-by-case basis; in some situations, a specific indication may actually alter the overall intended use of the device in which case the multiple predicates concept may not be applicable. More information regarding when a specific indication is reasonably included within a general indication can be found in FDA’s Guidance for Industry, “General/Specific Intended Use” (http://www.fda.gov/MedicalDevices/DeviceRegulationandGuidance/GuidanceDocuments/ucm073944.htm).
Features can also be added to a new device to increase convenience of use and/or functionality, without altering the intended use or risk profile (relative to a predicate) of the new device. Under such circumstances, the device with the added feature can be reviewed in a 510(k), even if the added feature consists of a component that may fall under a different classification regulation. A catheter-thermometer construct is useful in illustrating this concept.
Multiple Predicates Example 5:
A manufacturer submits a 510(k) for a urinary catheter with a thermometer. The thermometer/temperature-measuring feature is not affecting the intended use or risks of using the catheter (assuming it is integrated appropriately), nor is the catheter affecting the performance or risk profile of the thermometer. The temperature-measuring feature is a convenience component that is added to the catheter, with the intended use of the device still being that of the catheter to pass fluids to or from the urinary tract, so it is appropriate to have a legally marketed catheter serving as the primary predicate.
There are obvious limitations to feature/component additions in the 510(k) program. If a feature is added which alters the intended use of the new device and/or alters the safety profile (i.e., introduces new or additional risk factors) such that comparison to a predicate cannot be made, the new device is ineligible for the 510(k) program. A new device with a new design feature or added component must meet the SE standard with at least a single predicate from the same classification regulation.
-
Reference Devices When demonstrating substantial equivalence in a 510(k) submission, manufacturers sometimes direct attention to similar situations FDA has encountered in the past. If a manufacturer successfully navigates through Decision Point 4 on the Flowchart using a single predicate device, other legally marketed devices, which FDA calls “reference devices,” may be used to support scientific methodology or standard reference values at Decision Point 5a.
It is important to note that a reference device is not considered a predicate device and it cannot be used to address Decision Points 1 – 4 on the Flowchart. Additionally, the applicability of a reference device will need to be reviewed by FDA for its appropriateness. If a selected reference device is used in an anatomical location or for a physiological purpose that is considerably different than that of the new device, its utility as a reference device may be limited.
If a manufacturer intends to use a reference device, the manufacturer should provide a scientific rationale that justifies its use. This concept is illustrated in the Reference Device Examples below. We recommend that you read these examples side-by-side with the Flowchart in Appendix A so that you can follow the decision-making process.
Reference Device Example 1: A manufacturer submits a 510(k) for a total knee implant with coating X (the new device). Other coated knee implants with the same intended use with coatings A, B, and C are legally marketed. In addition, a total hip implant with coating X is legally marketed. The manufacturer cites the legally marketed knee implant with coating A as the predicate device. FDA determines that the new device has an appropriate predicate device (thus, answering “yes” at Decision Point 1) and the new device has the same intended use as the predicate device (thus, answering “yes” at Decision Point 2 in the Flowchart).16 However, FDA determines that the new device does not have the same technological characteristics as the predicate device (thus, answering “no” at Decision Point 3 in the Flowchart), because the new device (knee implant with coating X) has a chemical profile different from the chemical profile of the cited predicate device (knee implant with coating A). There are no other technological differences between the new device and the cited predicate device (knee implant with coating A). FDA determines that the new device does not raise different questions of safety and effectiveness. In this case, FDA determines that the safety and effectiveness questions regarding the coating material are whether it is biocompatible and whether it affects the fixation of the implant and these questions apply to both the new device and predicate device (thus, answering “no” at Decision Point 4 in the Flowchart).
After Decision Point 4 in the Flowchart, if appropriate, the manufacturer may refer to the reference device (the hip implant with coating X in this situation) to support the appropriate scientific methods for the characterization of coating X on the new knee implant device. In this particular example, the manufacturer provided an adequate scientific rationale to support that the methods used to characterize the biocompatibility and characteristics of the coating (e.g., strength, abrasion, etc.) on the hip implant are applicable to the knee implant.17 The reference device (hip implant with coating X) is used in this case solely to assist with the characterization of the coating on the new device (knee implant with coating X).
Reference Device Example 2: A manufacturer submits a 510(k) for an over-the-counter blood glucose test system (glucose meter). Other glucose meters with the same intended use are legally marketed. The manufacturer cites a legally marketed glucose meter as the predicate device. FDA determines that the new device has an appropriate predicate device (thus, answering “yes” at Decision Point 1) and the new device has the same intended use as the predicate device (thus, answering “yes” at Decision Point 2 in the Flowchart). The manufacturer has not demonstrated that the new device has the same technological characteristics as the predicate device (thus, answering “no” at Decision Point 3 in the Flowchart), but the new device does not raise different questions of safety and effectiveness (thus, answering “no” at Decision Point 4 in the Flowchart).
Because glucose meters of this type typically have relatively high inherent total error due to limitations in their technology and other factors, in order to sufficiently characterize the analytical performance of the new device (and answer “yes” at Decision Point 5a in the Flowchart), the new device uses the same approach to characterize analytical performance as the predicate device. Specifically, the accuracy of the new device is evaluated by comparing its blood glucose results to reference values generated on a laboratory-based glucose measurement device that has been well-validated for precision and accuracy, and that is traceable to a higher order, e.g., internationally recognized standard. If the performance of the new device (including accuracy compared to the reference values from a reference device) is equivalent to the performance of the predicate device (including accuracy of the predicate compared to the reference values from a reference device), the FDA would determine that the data demonstrate equivalence (thus answering “yes” at Decision Point 5b in the Flowchart).
- Lack of Predicate Device If a predicate device with the same intended use cannot be identified, or if the new device’s different technological characteristics raise different questions of safety or effectiveness, a manufacturer may submit a De Novo request, either after receipt of an NSE letter or directly requesting classification through the De Novo process.18 For high risk devices, a PMA (or alternative submission type) may be required.
- Identification and Documentation of the Predicate(s) Although manufacturers may cite more than one predicate device in a 510(k), FDA recommends that the manufacturer clearly identify the primary predicate device to which substantial equivalence is being claimed.19 Further, as part of the decision-making process, FDA should clearly cite the predicate device relied upon in determining substantial equivalence for the new device in its review documentation. If multiple predicates or reference devices are used in accordance with this guidance, the manufacturer should identify each device and explain why more than one predicate or a reference device is necessary and appropriate to support substantial equivalence. Manufacturers should choose the most appropriate single or primary predicate for their new device, and should limit the multiple predicates to those most helpful in facilitating review of the new device and to the minimum number necessary to support substantial equivalence. Predicate device(s) relied upon for SE must be accurately cited in the 510(k) Summary (see Appendix B) according to 21 CFR 807.92 (a)(3). Reference devices also may be cited in the 510(k) Summary.
D. Intended Use 🔗
Under section 513(i) of the FD&C Act, FDA may only determine that a device is substantially equivalent to a predicate device if it has the same intended use.20 (Refer to the 510(k) Decision-Making Flowchart in Appendix A). A finding of NSE due to a new intended use is relatively rare. Approximately 10% of all NSE decisions are due to a new intended use.21 This type of NSE determination generally reflects a finding that a change in the indications for use of a device creates a new intended use. This section of the guidance provides further clarification about the terms “intended use” and “indications for use,” describes how FDA determines what the intended use of a device is, and provides examples of changes in indications for use that may constitute a new intended use making the device ineligible for review under the 510(k) program.
-
Explanation of Intended Use and Indications for Use For purposes of substantial equivalence, the term intended use means the general purpose of the device or its function, and encompasses the indications for use. The term indications for use, as defined in 21 CFR 814.20(b)(3)(i), describes the disease or condition the device will diagnose, treat, prevent, cure or mitigate, including a description of the patient population for which the device is intended.22 The intended use of a device is one criterion that determines whether a device can be cleared for marketing through the 510(k) process or must be evaluated in a PMA (or alternative submission type), or if appropriate, a De Novo request. The proposed labeling in a 510(k) is used to determine a device’s intended use (Section 513(i)(1)(E) of the FD&C Act). The indications for use statement in a 510(k) is also a factor in determining a device’s intended use. Consistency between the indications for use statement and the proposed labeling will facilitate the review of the 510(k).
A finding of substantial equivalence means that the indications for use of the new device fall within the intended use of the predicate device and, therefore, the two devices have the same intended use. For devices with general indications for use that do not specify a disease, condition, or population (or an anatomical site from which a disease state or population may be inferred), the indications for use and intended use are the same. Such indications for use are referred to as “tool type” indications for use. Examples of devices with “tool type” indications for use include devices such as scalpels, which are often indicated for cutting tissue, or imaging devices, which are often indicated for taking images of the body. A scalpel indicated for removing a particular type of cancerous cell, however, has indications for use specific to the identified disease, condition, or population, and therefore, does not have “tool type” indications for use.
-
Determining Intended Use Section 513(i)(1)(E)(i) of the FD&C Act provides that the FDA’s determination of intended use of a device “shall be based upon the proposed labeling” submitted in a 510(k). When a review of the indications for use and all other information in the proposed labeling submitted with a 510(k) supports an intended use that is the same as that of the predicate device, FDA will determine that the new device and predicate device have the same intended use. This guidance does not address FDA’s authority to consider information outside the labeling in reviewing a 510(k) and issue an “SE with limitations” under section 513(i)(l)(E) of the FD&C Act because “there is a reasonable likelihood that the device will be used for an intended use not identified in the proposed labeling” and such use “could cause harm.” For information on “SE with limitations,” please see the guidance document, “Determination of Intended Use for 510(k) Devices; Guidance for CDRH Staff (Update to K98-1 )” (http://www.fda.gov/MedicalDevices/DeviceRegulationandGuidance/GuidanceDocuments/ucm082162.htm). When a review of the labeling submitted with a 510(k) shows that the indications for use of a new device and predicate device differ, FDA must evaluate whether the new23 indications for use fall within the same intended use as that of the predicate device. As described in Section IV.A, because the substantial equivalence determination is grounded in safety and effectiveness, this determination depends upon the safety and effectiveness of the new device for the new indications relative to the safety and effectiveness of the predicate device.
Once FDA has determined the indications for use of the new device upon review of the proposed labeling, FDA may rely upon relevant clinical and/or scientific information, that does not appear in the proposed labeling submitted with the 510(k), regarding the safety and effectiveness of the new indications for use. For example, FDA may rely upon publicly-available scientific information or Agency knowledge about how a disease progresses to determine whether indications for use to treat a certain disease or anatomical site constitute a new intended use.
-
Determining When Indications for Use Result in a New Intended Use Not every change in indications for use that may affect safety or effectiveness will result in a finding of a new intended use. Only a change in the indications for use that raises different questions of safety and effectiveness and therefore, precludes a meaningful comparison with the predicate device constitutes a new intended use. FDA may find changes in indications for use of a device to constitute a new intended use when the changes raise a safety or effectiveness issue that was not raised by the predicate device, or the changes have the potential to significantly increase a safety or effectiveness concern raised by the predicate device.24 In the first case, reliance on a predicate device is inadequate because the safety or effectiveness issue was not considered in reviewing the 510(k) for the predicate device. In the second case, although the safety or effectiveness issue may have been considered in the 510(k) for the predicate device, the finding of substantial equivalence for the predicate device cannot be generalized to the new indications for use because of a probable, significant change in the incidence or severity of the issue. In both cases, the predicate device is not an adequate “proxy” for an independent determination of safety and effectiveness.
Illustrative Example 1: A new device’s instructions for use describe using a general surgery device in a body cavity, but the predicate device is used only to treat external injuries. A comparison to the predicate device may not be adequate to address the risk of infection posed by internal use of the device. Because of the need for an independent assessment of an issue that was not evaluated or was of significantly less concern during FDA’s review of the 510(k) for the predicate device, FDA may determine that the indication for use of the new device constitutes a new intended use and a PMA (or alternative submission type), or if appropriate, a De Novo request, is required.
Illustrative Example 2: A 510(k) for an existing surgical ablation device cleared for ablation of cardiac tissue has now been submitted for the treatment of atrial fibrillation. While the devices are similar in technology, additional clinical testing has been conducted to demonstrate that not only can the device ablate cardiac tissue, but also that doing so can treat atrial fibrillation safely and effectively. While the question of whether or not cardiac tissue can be safely and effectively ablated was raised by the predicate device, FDA has determined that the specific indication for the treatment of atrial fibrillation constitutes a new intended use because it raises questions of both safety and effectiveness not raised by the predicate device. Specifically, treatment of atrial fibrillation requires extensive ablation to create linear lines of conduction block in a maze-like pattern that eliminates fibrillatory conduction in the atria. The effectiveness assessment for the treatment of atrial fibrillation warrants a clinical outcome study. Furthermore, the risks of iatrogenic heart block and collateral cardiac or extra-cardiac damage are either raised or increased when such a complex and extensive lesion set is created. As a result, a PMA (or alternative submission type) is required.
-
Changes in Indications for Use that May Result in a New Intended Use All new indications for use should be evaluated to determine whether they reflect a new intended use. Certain types of changes, however, warrant particular attention in evaluating whether the new indications for use result in a new intended use because they are more likely to significantly affect safety or effectiveness:
- a change from a functional/performance indication to a treatment or aesthetic indication;
- a change from a diagnostic indication to a screening indication, or vice versa;
- a change in the anatomical structure of use;
- a change in the patient population (e.g., adult versus pediatric; different disease populations);
- a change in the clinical context or setting (e.g., periodic monitoring versus continuous monitoring; hospital versus home use).
E. Technological Characteristics 🔗
After FDA has determined that a valid predicate device exists for a new device and that both devices have the same intended use, FDA will move to Decision Points 3 and 4 of the Flowchart (see Appendix A). In these steps of the 510(k) review process, FDA compares the technological characteristics of the new device and the predicate device to determine whether the new device has the same technological characteristics as the predicate, and if not, whether the different technological characteristics raise different questions of safety and effectiveness.25 Devices reviewed under the 510(k) program commonly have different technological characteristics from their predicate device(s); however, FDA rarely makes a finding of NSE at Decision Point 4.26
-
Step 1 – Identification of Technological Characteristics of the New and Predicate Device For FDA to evaluate whether differences exist between the technological characteristics of the new device and the predicate device(s), the manufacturer should clearly identify the technological characteristics of each device individually. Technological characteristics include materials, design, energy source, and other device features, as defined in section 513(i)(1)(B) of the FD&C Act and 21 CFR 807.100(b)(2)(ii)(A).
To facilitate FDA’s review of a device’s technological characteristics, the device description in a 510(k)27 should include the information necessary to explain the new device’s technological characteristics, including similarities in materials, design, energy source, and other device features. This information will be evaluated by FDA to determine whether the technological characteristics of the new device are different and, if so, whether they raise different questions of safety and effectiveness as compared to the predicate(s). Examples of key characteristics that should be provided as part of a 510(k) submission include, but are not limited to, the following features:
- An overall description of the device design. A complete description of the device may be facilitated by the submission of engineering drawings or other figures. If the device consists of multiple components, a diagram identifying how the different components of the device system work together may be beneficial. The device description should also include a discussion of the physical specifications, dimensions and design tolerances that are critical to the new device.28 Significant features of the new device should have a clear purpose within the context of the overall design and intended use. In cases where this is not apparent, it is important for the 510(k) submission to provide a discussion of how a particular device design or component contributes to the overall use and function of the new device.
- Materials. For many devices, a complete identification of the detailed chemical formulation used in the materials of construction, especially for those materials that come into contact with the patient, should be provided. Note that the FDA does not clear/approve materials.29 Any additives, including color additives, coatings, or other surface modifications should also be identified. For some devices, the processing of the material (e.g., forged vs. cast) or the state of the material (e.g., amorphous vs. crystalline) may also significantly contribute to or affect the overall safety or function of the device, and so should be included as part of the device description, as applicable.
- Energy sources. This not only includes energy delivery to the device, including the use of batteries, but also energy delivery that is part of the functional aspect of the device (e.g., laser, radiofrequency, ultrasound, etc.) and that affects the patient and/or the health care professional using the device. Where applicable, a discussion of this characteristic should be provided.
- Other key technological features. These include, but are not limited to, software/hardware features, density, porosity, degradation characteristics, nature of reagents (recombinant, plasma derived, etc.), principle of the assay method, etc., that are not explicitly included as part of the materials, design or energy source characteristics. These technological features should be included as part of the device description in the 510(k) submission, as appropriate for the specific device technology.30
A 510(k) submission must also contain information about the technological characteristics of the predicate device (21 CFR 807.87(f), 807.92(a)(3) and (a)(6)). The manufacturer of the new device should provide information necessary and sufficient to fully and clearly identify and describe the technological characteristics of the predicate device so that FDA can conduct a comparative assessment of the technological characteristics, as further described in Step 2.
-
Step 2 – Identification of Differences in Technological Characteristics Between the New and Predicate Device Once the technological characteristics of the new and predicate device(s) have been clearly identified, the next step involves a comparison of these characteristics to identify any differences. This may involve a comparison of detailed specifications as well as a comparison of the system-level technological characteristics of the devices. FDA relies upon information provided about the predicate device, in addition to the information in our files as appropriate, and the new device to determine whether the new device has different technological characteristics (Decision Point 3) in comparison to the predicate(s).
At this point, FDA will assess whether the similarities/differences in technological characteristics between the new and predicate device(s) have been appropriately identified. FDA highly recommends that the manufacturer summarize this information in tabular format to facilitate this step of review.
-
Step 3 – Determination of Whether the Differences in Technological Characteristics Raise Different Questions of Safety and Effectiveness If FDA determines that there are differences in the technological characteristics of the new device and the predicate device, FDA will review and evaluate all relevant information bearing on any such differences in technological characteristics to determine whether they raise different questions of safety and effectiveness for the new device as compared to the predicate device (Decision Point 4 on the Flowchart). A “different question of safety or effectiveness” is a question raised by the technological characteristics of the new device that was not applicable to the predicate device, and poses a significant safety or effectiveness concern for the new device.
Some examples are provided below to illustrate cases where the response to this general question was “yes,” i.e., the new device was determined to raise different safety and effectiveness questions in comparison to the predicate device, and the new device was found NSE.
Illustrative Example 1
Predicate: A biological indicator utilizing natural bacterial spores with recognized resistance characteristics as organisms for the biological indicator, where the presence of a color change or fluorescent signal is indicative of bacterial viability.
New Device: A biological indicator based on recombinant technology/genetic engineering, where the fluorescent signal is not indicative of bacterial viability; it is indicative of plasmid enzyme expression.
Intended Use: Same
Different questions of safety and effectiveness? Yes
Why: Due to the engineering of a plasmid into the biological indicator, it is possible to have viable bacteria that do not contain the plasmid in sufficient amounts to generate a signal. In this case, the biological indicator could falsely indicate that the monitored load was sterilized properly due to the absence of fluorescent signal, while there are viable non-expressing bacteria in the indicator (development of false negatives). This technological difference raises different types of safety and effectiveness concerns for using a recombinant-DNA plasmid that codes for antibiotic resistance and a signaling enzyme in the spores of a biological indicator. Appropriate test methodologies and risk assessments need to be determined to address the properties of the introduced plasmid and host bacterial spore that could affect indicator performance. Because these types of questions were not necessary to take into account for the predicate device, the new device would be found NSE.
Illustrative Example 2
Predicate: A mechanical device used for embryo dissection
New Device: An electrical device used for embryo dissection
Intended Use: Same
Different questions of safety and effectiveness? Yes Why: In this example, changing the process from a mechanical process to an electrical energy source (e.g., laser) changes the way the device operates and raises different safety concerns regarding how the heating aspect of the electrical mechanism affects the embryo. Because these types of questions were not necessary to take into account for the predicate device, the new device would be found NSE.
Illustrative Example 3
Predicate: A device inserted into the patient’s pharynx through the mouth to provide a patent airway by mechanically moving the soft tissue.
New Device: A device placed externally on the mandible and neck to apply a vacuum to move the soft tissue forward and thus “open” the airway.
Intended Use: Same
Different questions of safety and effectiveness? Yes
Why: The predicate device is invasive and placed midline in the oropharynx and does not exert pressure on the vascular, respiratory, or nerve structures in the neck, whereas the new device exerts continuous external negative pressure on these areas, raising different types of safety questions, such as the risks and potential adverse events associated with the stimulation of the nerve structures in the neck. Because these types of questions were not necessary to take into account for the predicate device, the new device would be found NSE.
In the event the answer to Decision Point 4 is “No” and the differences between the new device and 21 the predicate device do not raise different questions of safety and effectiveness, then the scientific review of the performance data will proceed. However, if the answer to Decision Point 4 is “Yes” and the differences between the new device and predicate device raise different questions of safety and effectiveness, then the new device will be found NSE. Upon receipt of this type of NSE letter, the manufacturer may submit a PMA (or alternative submission type), or if appropriate, a De Novo request.
F. Requests for Performance Data 🔗
Although FDA may rely upon descriptive information alone to address the critical questions in the Flowchart (Decision Points 1 through 4), performance data are typically needed in a Traditional 510(k) to demonstrate the substantial equivalence of a new device to a predicate device. In addition, information on device performance described in labeling or other sections of the 510(k) should be supported with appropriate performance data. The type and quantity of performance data necessary to support a determination of substantial equivalence depend upon the device and/or device type.31 Performance data may be needed to address a variety of safety and effectiveness issues and may be generated from different types of tests and studies.
FDA’s data requests typically follow a stepwise analytical process to ensure the information requested reflects the least burdensome approach to establishing substantial equivalence.32 First, FDA considers whether descriptive information about the technological characteristics, such as the materials, design, and specifications, of the new device is sufficient. Very few 510(k) submissions rely solely on descriptive information about materials, design, specifications, and other technological characteristics (see 21 CFR 807.87(f) and (g)). When this information is not sufficient to support a substantial equivalence determination, FDA then considers whether non-clinical bench performance testing or analytical studies using clinical samples would be sufficient. For in vitro diagnostic devices (IVDs), analytical studies include, but are not limited to, evaluations of accuracy, precision, specificity, and sensitivity. Non-clinical bench performance testing includes a wide variety of test modalities that will be dependent upon the specifics of the actual device, including, but not limited to:
- mechanical, electrical, and biological engineering performance, such as fatigue, wear, tensile strength, compression, flowrate, burst pressure;
- electromagnetic compatibility (EMC);
- sterility;
- stability/shelf life;
- software validation;
- other forms of non-clinical, including device-specific.
Non-clinical animal and/or biocompatibility studies are typically requested when other forms of nonclinical bench performance testing are not sufficient to demonstrate substantial equivalence. Nonclinical laboratory studies that support the safety of medical devices must be conducted in compliance with 21 CFR Part 58, Good Laboratory Practice (GLP) for Nonclinical Laboratory
Studies, as applicable, to ensure the quality, reliability, and integrity of study data.33 For more information on this topic, see FDA’s Draft Guidance for Industry and Food and Drug Administration Staff, “The Applicability of Good Laboratory Practice in Premarket Device Submissions: Questions & Answers” (http://www.fda.gov/medicaldevices/deviceregulationandguidance/guidancedocuments/ucm366338.htm). FDA’s draft guidance represents FDA’s proposed approach on this topic.
When analytical or non-clinical bench performance testing data, or non-clinical animal and/or biocompatibility studies are insufficient, or available scientific methods are not acceptable, e.g., the scientific methods are deemed unacceptable because they are not clinically validated or are not supported by a valid scientific rationale, FDA may request clinical performance data to support a substantial equivalence determination. For 510(k)s reviewed in the Office of Device Evaluation, FDA currently requests clinical data for less than 10 percent of the 510(k) submissions. In some instances, clinical data may be a less burdensome means of demonstrating substantial equivalence than other means of performance testing, and 510(k)s reviewed in CBER for products intended to ensure the safety and effectiveness of blood and blood products typically include clinical data. Clinical data provided in support of any marketing application, including a 510(k) when those data are relevant to a substantial equivalence determination, should constitute valid scientific evidence as defined in 21 CFR 860.7(c)(2)34 and must comply with the Investigational Device Exemptions (IDE) regulations as applicable.35
Although not an exhaustive list of instances in which FDA may request clinical data to demonstrate substantial equivalence,36 the following scenarios illustrate the most common situations in which clinical data may be requested. As explained in the Scope Section (see Section III), the information in this guidance and the examples below do not take the place of any device-specific guidance.
Note: The examples provided below distinguish between examples that are only applicable to diagnostic devices, including IVDs, and therapeutic devices. This is because there are significant differences in the clinical data requirements for these two categories of devices.
-
New or Modified Indications for Use – Same Intended Use In rare instances, FDA may rely upon clinical data to determine that new or modified indications for use fall within the same intended use as a predicate device.
Illustrative Examples:
- The new device is an IVD that is indicated for over-the-counter use, whereas the predicate device is indicated for prescription use in the home or prescription use in a clinical setting. The newly indicated test population might fall within the intended use of the predicate device. Clinical data (demonstrating that the user can collect the sample, generate an accurate result, and adequately interpret the result) might establish that the indication for use for the new device falls within the intended use of the predicate device.
- The new IVD is indicated for use with patients who have symptoms and signs of illness from any member of a specified set of closely related diseases. The indications for use for the predicate IVD do not include one of the diseases addressed by the new IVD. Clinical data (concerning all diseases in the newly specified set) might establish that the indications for use for the new device fall within the intended use of the predicate device.
- The manufacturer modifies the indications for use, explicitly or implicitly, by proposing a different surgical implantation method which also affects the indications for use, e.g., a minimally invasive procedure in place of an open procedure, and the safety and effectiveness of the new device cannot be adequately replicated or otherwise characterized in a non-clinical performance (including animal) test environment to adequately support substantial equivalence to the predicate. Although on its face a minimally invasive procedure would appear to involve less serious risks than an open procedure, the minimally invasive procedure may be less effective or may present different but still serious risks.
-
Technological Differences FDA may request clinical data for a 510(k) when the technological differences between the new device and predicate device are significant but do not support an immediate NSE determination due to different questions of safety and effectiveness. In these limited situations, clinical data may be needed to evaluate the safety and effectiveness of the new device as compared to the predicate device.
Illustrative Examples:
- A new IVD uses the same analyte-specific chemistry as the predicate, but with a different read-out technology (e.g., chemiluminescence instead of colorimetry). Clinical data may be necessary to demonstrate that the new device performs equivalent to the predicate.
- Performance characteristics of the new device in comparison to the predicate are significantly different in non-clinical performance testing, e.g., the predicate is rigid whereas the new device is designed to be more flexible. Clinical data may be necessary to demonstrate that the new device performs equivalent to the predicate.
- Some devices that display data about the patient’s anatomy or physiology, e.g., glucose meters, pulse oximeters, blood pressure cuffs, are supported by software. If there is a change in the software that relates to how the software analyzes the patient’s anatomy or physiology, the device may need to be tested on actual patients to assure that the software performs in a manner that is equivalent to the previous version. In this case, non-clinical data may not suffice.
- The technological characteristics of the new device raise a question concerning whether its clinical performance can be expected to be equivalent to the clinical performance of the predicate. Clinical data may be necessary to demonstrate that the new device performs equivalent to the predicate. For IVDs, an example is a new prothrombin time (clotting) test using thromboplastin that is a recombinant product instead of a naturally occurring material.
-
Non-clinical Testing Methods are Limited or Inappropriate Because of the Indications for Use or Device Technology FDA requests clinical data for a 510(k) submission to address issues that cannot be adequately addressed using non-clinical test methods because of the indications for use or device technology. For instance, for certain indications or technologies, FDA may request clinical data when non-clinical testing methods are not validated, are limited or are inappropriate, because of either their scope or their applicability, to demonstrate substantial equivalence.
Illustrative Examples:
- For some devices, the way they are used and the environment in which they are used affect the way they perform. For example, the non-clinical performance testing on the new device may be insufficient to support a substantial equivalence determination if the testing cannot replicate the way the device will be used or the way similar devices have been demonstrated to fail in a clinical setting. Although the non-clinical testing for these devices might be informative for many other aspects of the device, it may be necessary to supplement the nonclinical data with clinical simulation performance data or clinical performance data.
- If the non-clinical testing of a device raises safety concerns that cannot be mitigated or answered through non-clinical testing, such a device may require clinical testing to assure that the safety questions are not greater than those raised by the predicate device.
New scientific information may affect FDA’s expectations concerning the type and level of performance data included in a 510(k) submission. For device types with long histories of safe use and well understood mechanisms of action, more limited performance testing data may be sufficient. On the other hand, a pattern of adverse events or published literature documenting poor clinical outcomes with a particular technology may lead FDA to reconsider its regulatory approach to premarket submissions for such technology.37 Should FDA change its scientific decision making with regard to a particular device, FDA will consider its options (e.g., guidance, advisory panel meeting, etc.), for explaining such change and the basis for the decision to ensure transparency in the change in policy. FDA also intends to consider any pending 510(k) submissions that may be affected by the change or allow for an appropriate transition period, in certain situations that may affect the industry at large.
G. The 510(k) Summary 🔗
The 510(k) Summary38 is a document that provides a high-level discussion of the content of a 510(k) and must include all the elements identified in 21 CFR 807.92. A 510(k) Summary must be in sufficient detail to provide an understanding of the basis for a determination of substantial equivalence (21 CFR 807.92(a)).
In an effort to improve the transparency and predictability of the 510(k) program and to ensure that the 510(k) Summary reflects the information provided in a 510(k) submission to support a substantial equivalence determination, FDA intends to verify the accuracy and completeness of the information included in a 510(k) Summary.
Although the 510(k) Summary is a document created by the manufacturer and is included in the 510(k), revisions to the 510(k) Summary may be necessary to accurately reflect the FDA’s decision-making process. For example, manufacturers may have identified several devices as potential predicate devices, whereas, in the course of FDA’s substantial equivalence evaluation, FDA may have determined that only one of these devices is an appropriate predicate device. In addition, it is possible during the course of FDA’s review of the 510(k), that additional information or testing may be requested and submitted. Consequently, the manufacturer may be requested to update the 510(k) Summary to accurately include and convey the information identified in 21 CFR 807.92 and which was used to support the final decision-making process.
In Appendix B, FDA describes the requirements of the content to be included in a 510(k) Summary, in accordance with 21 CFR 807.92, and provides guidance on the information to be included in a 510(k) Summary to ensure compliance with 21 CFR 807.92 and consistency in the level of information conveyed and captured in the 510(k) Summaries which are available to the public on FDA’s website. In Appendix C, FDA has provided a hypothetical 510(k) Summary in order to demonstrate the recommended level of detail for each section.
Appendix A. 510(k) Decision-Making Flowchart 🔗
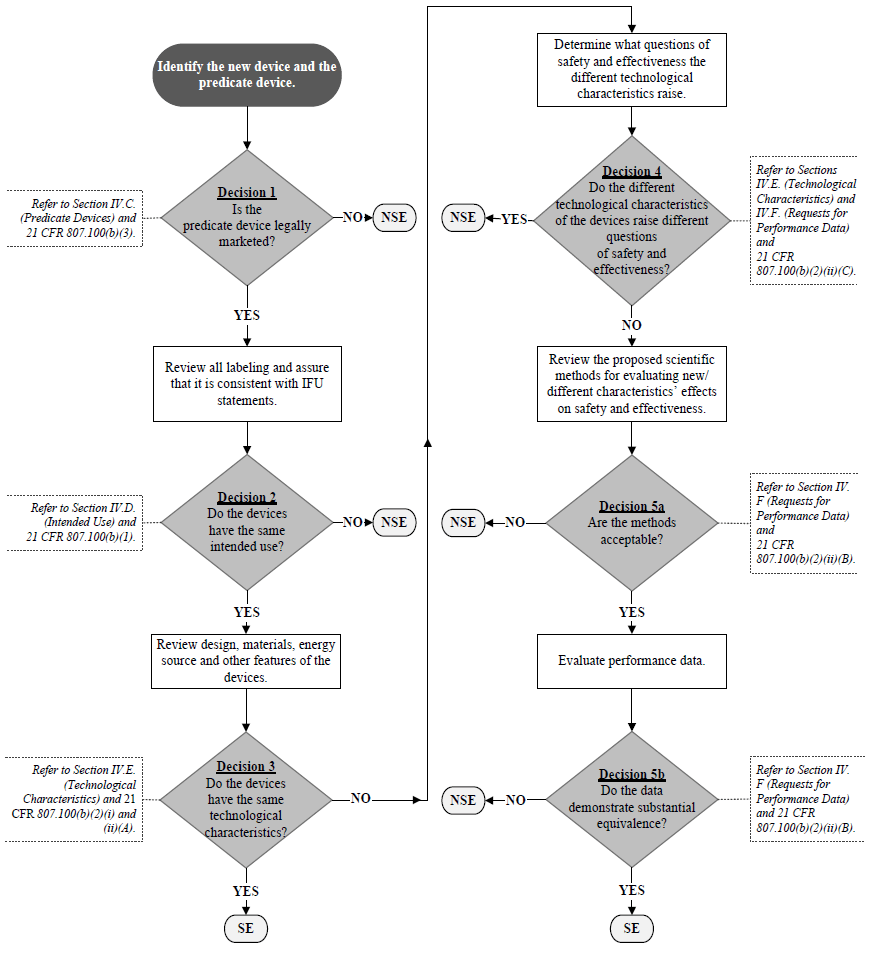
SE = “Substantially Equivalent”
NSE = “Not Substantially Equivalent”
IFU = “Indications For Use”
This Flowchart is not intended to be used as a ‘stand-alone’ document and should only be considered in conjunction with the accompanying text in this guidance.
Appendix B. The 510(k) Summary Document Requirements 🔗
In Appendix B, FDA provides further clarification and guidance to facilitate compliance with the requirements set forth in 21 CFR 807.92 and consistency in the information conveyed in the 510(k) Summaries which are available to the public on FDA’s website. As noted earlier in this guidance document, if during the course of review, additional testing or information are requested, the manufacturer should submit a revised 510(k) Summary to reflect the additional information. The following identifies the information that must be included in the 510(k) Summary under 21 CFR 807.92, information that we recommend be included in the 510(k) Summary, and other considerations.
- 807.92(a)(1): “The submitter's name, address, telephone number, a contact person, and the date the summary was prepared.”
- The “submitter” or manufacturer should be the holder of the 510(k), not a consultant or law firm.
- 807.92(a)(2): “The name of the device, including the trade or proprietary name if applicable, the common or usual name, and the classification name, if known.”
- FDA recommends that the manufacturer list all applicable names and model numbers, if known.
- If the submission is bundled39, the 510(k) Summary should list all applicable classification regulations and product codes.
- 807.92(a)(3): “An identification of the legally marketed device to which the submitter claims equivalence. A legally marketed device to which a new device may be compared for a determination regarding substantial equivalence is a device that was legally marketed prior to May 28, 1976, or a device which has been reclassified from class III to class II or I (the predicate), or a device which has been found to be substantially equivalent through the 510(k) premarket notification process.”
- FDA recommends that the manufacturer provide the 510(k) number of the device used as the predicate device in support of the current 510(k) submission.
- If using an exempt device as a predicate, the manufacturer should list the classification regulation and the product code.
- If using a device that has been reclassified from Class III to II as a predicate, where a 510(k) has not been submitted, please list the PMA number.
- If the manufacturer lists an inappropriate predicate device, FDA will request that such information be removed and the 510(k) Summary updated accordingly by the manufacturer.
- 807.92(a)(4): “A description of the device that is the subject of the premarket notification submission, such as might be found in the labeling or promotional material for the device, including an explanation of how the device functions, the scientific concepts that form the basis for the device, and the significant physical and performance characteristics of the device, such as device design, material used, and physical properties.”
The description of the device attributes should include the following details:- Device Identification:
- List all key device components included in the submission (e.g., catheter, cable wire, leads)
- List all model numbers (if known) and briefly explain the differences among models
- Device Characteristics (address all that apply):
- software
- biologics
- drugs
- any patient-contacting materials
- coatings
- additives
- single-use
- sterile
- sterilization method [specify]
- Environment of Use (address all that apply):
- healthcare facility/hospital
- home
- other [specify]
- Brief Written Description of the Device:
- Explanation of how the device works/principle of operation
- Mechanism of action
- Any necessary feature to determine SE or device performance
- Energy source (if applicable)
- Materials of Use
- General type of material used (e.g., polysulfone, stainless steel)
- If material conforms to an FDA recognized consensus standard for medical use, include the applicable number (e.g., ASTM FXXXX-last 2 numbers of the year)
- Duration and type of contact
- Key Performance Specifications/Characteristics of the Device
- Device Identification:
- 807.92(a)(5): “A statement of the intended use of the device that is the subject of the premarket notification submission, including a general description of the diseases or conditions that the device will diagnose, treat, prevent, cure, or mitigate, including a description, where appropriate, of the patient population for which the device is intended. If the indication statements are different from those of the legally marketed device identified in paragraph (a)(3) of this section, the 510(k) summary shall contain an explanation as to why the differences are not critical to the intended therapeutic, diagnostic, prosthetic, or surgical use of the device, and why the differences do not affect the safety and effectiveness of the device when used as labeled.”
- The 510(k) Summary should include the Indications for Use, which should be identical to that proposed on the Indications for Use Sheet and the labeling.
- If the Indications for Use are different from those of the predicate device, a brief explanation is required to address why the differences in the Indications do not affect the safety and effectiveness of the device and do not alter the intended therapeutic, diagnostic, prosthetic, or surgical use of the device.
- 807.92(a)(6): “If the device has the same technological characteristics (i.e., design, material, chemical composition, energy source) as the predicate device identified in paragraph (a)(3) of this section, a summary of the technological characteristics of the new device in comparison to those of the predicate device. If the device has different technological characteristics from the predicate device, a summary of how the technological characteristics of the device compare to a legally marketed device identified in paragraph (a)(3) of this section.”
- 807.92(b): “510(k) summaries for those premarket submissions in which a determination of substantial equivalence is also based on an assessment of performance data shall contain the following information:”
“(1) A brief discussion of the nonclinical tests submitted, referenced, or relied on in the premarket notification submission for a determination of substantial equivalence,”
- A high level summary of the tests that were used to demonstrate substantial equivalence should be included (e.g., fatigue testing, biocompatibility, etc.).
- If a guidance document was referenced/used for the testing, the guidance document should be referenced in this section.
- If an FDA recognized consensus standard (e.g., test method or guide) was used/relied upon for testing, please list the standard connotation (e.g., ASTM FXXXX-last 2 numbers of the year).
“(2) A brief discussion of the clinical tests submitted, referenced, or relied on in the premarket notification submission for a determination of substantial equivalence. This discussion shall include, where applicable, a description of the subjects upon whom the device was tested, a discussion of the safety or effectiveness data obtained from the testing, with specific reference to adverse effects and complications, and any other information from the clinical testing relevant to a determination of substantial equivalence,”
- FDA is interested in collecting an appropriate degree of detail within this section to be informative regarding the level of evidence that was necessary to support an SE determination.
- As applicable, FDA recommends the following details be included regarding the clinical evidence provided to support an SE determination:
- Level of Evidence (identify one)
- Randomized, multi-arm, “blinded” study with concurrent sham (placebo) control
- Randomized, multi-arm, “blinded” study with concurrent (“active”) control
- Randomized, multi-arm, un”blinded” study with a control (control that is either active or consists of no treatment)
- Non-randomized study with concurrent (“active”) control
- Single-arm study with patient serving as own control (include designed singlearm crossover)
- Single-arm study with Historical Control (using patient-level data)
- Single-arm study with Literature Control (historical control)
- Single-arm study with Objective Performance Criteria
- Single-arm study with Performance Goals
- Registry
- Observational study
- Systematic review (meta-analysis with patient-level data)
- Meta-analysis based on summary information only
- Literature Summary
- Uncertain
- Location of Study (specify one of the following)
- United States only
- outside of United States only
- both in United States and outside of United States
- Identify applicable IDE number [Gxxxxxx]
- Primary Safety Endpoint Identified?
- If Yes, describe
- Primary Effectiveness Endpoint Identified?
- If Yes, describe
- Primary Composite Safety/Effectiveness Endpoint Identified, if applicable?
- If Yes, describe
-
Patient Accountability (Enter number of patients reported at each stage):
Stage Investigational Device Arm Total Control Arm Total Total Enrollment Treatment Primary Safety Endpoint Analysis Primary Effectiveness Endpoint Analysis Primary Composite Safety/Effectiveness (if app) The content of the table may need to be modified depending upon the specifics of the clinical data provided and the endpoints studied.
- Identify whether the study met the primary endpoint
- Whether Yes or No, describe
- Describe the study results in appropriate parameters
- Identify the adverse events and complications observed in the study, including those associated with the device.
“(3) The conclusions drawn from the nonclinical and clinical tests that demonstrate that the device is as safe, as effective, and performs as well as or better than the legally marketed device identified in paragraph (a)(3) of this section.”
- A brief summary of why the device is substantially equivalent to the predicate.
- Level of Evidence (identify one)
- 807.92(c): “The summary should be in a separate section of the submission, beginning on a new page and ending on a page not shared with any other section of the premarket notification submission, and should be clearly identified as a ‘510(k) summary’.”
- 807.92(d): “Any other information reasonably deemed necessary by the agency.”
- If the FDA determines that other information needs to be included within the 510(k) Summary, such information must be included within this document.
Appendix C. Sample of 510(k) Summary Complying with 21 CFR 807.92 🔗
510(k) Summary
I. SUBMITTER
Device Submitter, Inc.
123 Main Street
Anywhere, MD 01234
Phone: 555-555-1234
Fax: 555-555-0123
Contact Person: John Contact
Date Prepared: May 16, 2013
II. DEVICE
Name of Device: Brand X Endoscopic Stapling System, Model x123, Model y456
Common or Usual Name: Endoscopic Stapling
System Classification Name: Endoscope and Accessories (21 CFR 876.1500)
Regulatory Class: II
Product Code: ODE
III. PREDICATE DEVICE
Brand Z Endoscopic Plication System, KXXXXXX
This predicate has not been subject to a design-related recall.40
No reference devices were used in this submission.
IV. DEVICE DESCRIPTION
The Brand X Endoscopic Stapling System consists of a flexible endoscope, an endoscopy suite, and a number of associated accessories. The endoscope and staples are provided sterile (EtO).
Brand X uses an implant (surgical staple) that is delivered by a flexible endoscope by a surgeon or gastroenterologist for approximating adjoining portions of the esophageal and gastric tissues at the gastroesophageal junction, thereby creating a permanent surgical fundoplication. The system includes an ultrasonic transducer that operates as a range finder for measuring the relative alignment and the distance between the transducer at the tip of the endoscope (the anvil) and the ultrasonic mirror in the cartridge (that includes the staples). The device deploys sets of implantable staples for performing the fundoplication procedure.
The system combines a video camera, an ultrasonic range finder and a surgical stapler in a single unit. The flexible endoscope includes light guides, irrigation, air insufflation and suction channels that terminate at the endoscope tip. The stapler portion includes a cartridge that contains sterile 4.8 mm standard "B" shaped titanium surgical staples. Model x123 includes five (5) staples per cartridge, while Model y456 contains only four (4). The tip of the endoscope contains an anvil for the staples, as well as two small stainless steel screws that are extracted from the tip of the endoscope and engage into nuts positioned in the cartridge. The ultrasonic range finder measures the distance between an ultrasonic mirror in the cartridge and the tip of the endoscope. It also verifies alignment between the cartridge and the anvil, before insertion of the screws.
The endoscopy suite includes the ISL (Insufflation, Suction and Light) console, the CCU (Camera Control Unit) console and a monitor. The ISL console provides suction, insufflation and a white light (Xenon) source for illumination. The CCU console contains a controller for the camera, ultrasonic range finder and sensors that indicate status of the bending angle, screws and fire. The monitor displays patient information, the video image, and the processed data from the controllers such as ultrasonic data, fire status, degree of bending and screw position. A keyboard for entering data during the procedure is also included. The System includes three software applications: the video controller software, the ultrasound controller software, and the ISL controller software. The software systems work in conjunction with the hardware consoles listed above in order to visualize the procedure and deliver the staples. The endoscope is designed for single-patient use, and it is connected to the CCU and ISL consoles via a multi-connector. The endoscope handle contains the controls used by the operator to manipulate the endoscope.
The associated accessories include:
- Irrigation bottle with liquids for irrigation of the camera lens
- Suction canister for extracting liquids during the procedure
- Silicon tubes for connecting the ISL and other accessories to the endoscope
- Disposable air filter of the suction ISL input channel
- Overtube for protecting patient's pharynx
V. INDICATIONS FOR USE
The Brand X Stapling System is indicated for the endoscopic placement of surgical staples in the soft tissue of the esophagus and stomach in order to create anterior partial fundoplication for treatment of symptomatic chronic Gastro Esophageal Reflux Disease (GERD) in patients who require and respond to pharmacological therapy.
The Indications for Use statement for the Brand X device is not identical to the predicate device; however, the differences do not alter the intended therapeutic use of the device nor do they affect the safety and effectiveness of the device relative to the predicate. Both the subject and predicate devices have the same intended use for the treatment of GERD, by approximating tissue in the esophagus and stomach.
VI. COMPARISON OF TECHNOLOGICAL CHARACTERISTICS WITH THE PREDICATE DEVICE
Transoral endoscopic fundoplication is the technological principle for both the subject and predicate devices. It is based on the use of endoscopic instrumentation for approximating and permanently adjoining gastric and esophageal tissues, creating plications at the level of the gastroesophageal valve and thereby restoring valvular functionality and reducing gastric reflux into the esophagus. At a high level, the subject and predicate devices are based on the following same technological elements:
- Endoscope – used to reach the target tissue
- Device inserted through an overtube – to protect the esophagus
- Creation of a gastric (or gastroesophageal) plication in close proximity to the gastroesophageal junction by the retroflexed device
- Use of a permanent implant to secure tissue
- Use of a mechanical component for positioning and launching the implant
- User-controlled mechanical trigger (or knob) to launch the fastener (implant)
- Mechanically securing the plication by a permanent implant fastener
The following technological differences exist between the subject and predicate devices:
- Use of an ultrasound range finder
- Use of a staple as a fastener
- Use of different tissue capture and fixation mechanisms
- The predicate device must be used in conjunction with a flexible endoscope whereas the subject device has a flexible endoscope incorporated into the system.
VII. PERFORMANCE DATA
The following performance data were provided in support of the substantial equivalence determination.
Biocompatibility testing
The biocompatibility evaluation for the Brand X device was conducted in accordance with the FDA Blue Book Memorandum #G95-1 “Use of International Standard ISO-10993, ‘Biological Evaluation of Medical Devices Part 1: Evaluation and Testing,’” May 1, 1995, and International Standard ISO 10993-1 “Biological Evaluation of Medical Devices − Part 1: Evaluation and Testing Within a Risk Management Process,” as recognized by FDA. The battery of testing included the following tests:
- Cytotoxicity
- Sensitization
- Irritation
- Systemic toxicity
- Pyrogen Testing
The endoscopic delivery system is considered tissue contacting for a duration of less than 24 hours, while the staples are considered permanent implants. The titanium staple material conforms to ASTM F-67-06 for chemical composition.
Electrical safety and electromagnetic compatibility (EMC)
Electrical safety and EMC testing were conducted on the Brand X device, consisting of the ISL console, CCU console and endoscope. The system complies with the IEC 60601-1, IEC 60601-2-18 and IEC 60601-2-37 standards for safety and the IEC 60601-1-2 standard for EMC.
Software Verification and Validation Testing
Software verification and validation testing were conducted and documentation was provided as recommended by FDA’s Guidance for Industry and FDA Staff, “Guidance for the Content of Premarket Submissions for Software Contained in Medical Devices.” The software for this device was considered as a “major” level of concern, since a failure or latent flaw in the software could directly result in serious injury or death to the patient or operator.
Mechanical and acoustic testing
- Acoustic Testing
- Elongation of the bending cable
- Torque on the handle wheel and force on cable
- Crimp assembly, cable tensile strength, cable flexibility, minimum bending radius of the cables
- Staple verification
- Simulated use testing
Animal Study
In the animal study conducted, 16 pigs underwent endoscopy with the Brand X System. Twelve pigs underwent fundoplication, and 4 pigs served as a sham (control) group. There were no procedure related complications or premature deaths in this study, at the 2, 4 and 6 week follow-up (4 pigs in each group).
The safety and feasibility of the Brand X device were evaluated by macroscopic and histological evaluation of the tissue in the treatment stapled areas. These studies demonstrated that the Brand X device can safely create an anterior partial fundoplication, similar to that which is constructed using other endoscopic suturing devices.
Clinical Studies
Clinical testing of the Brand X device included an initial feasibility study of 6 patients, a pilot study consisting of 13 patients and a pivotal study of 72 patients. Substantial equivalence was based in part on the pivotal study.
Pivotal Study
The pivotal study was a prospective, multi-center, open label, non-randomized, single arm study of 72 patients, of which 66 were available for primary endpoint analysis; 3 subjects did not complete the procedure, and 3 were excluded from the effectiveness analysis. The device was used to staple the fundus of the stomach to the esophagus, using standard B shaped surgical staples. Stapling was performed in two or three locations at least 1.5 cm above the GE junction, separated by at least 90 degrees. The procedure was intended to create a partial anterior fundoplication as a reflux barrier. Patients were followed for a period of six months at 6 sites both in the United States and outside of the United States under IDE G070136.
Primary effectiveness endpoint:
GERD health related quality of life score (GERD-HRQL) off proton pump inhibitor (PPI) was improved from baseline by > 50%, at six months post procedure in at least 53% of the patients (53% is the lower boundary of the 95% confidence interval).
Primary safety endpoint:
The primary safety endpoint consisted of all treatment-related adverse events, during and after the procedure. “Treatment-related” events were conventionally defined as those which occurred in the first 30 days post-procedure.
Effectiveness
The primary endpoint for the Brand X study focused on the GERD-HRQL score. The study results demonstrated that 75% of the patients had a >50% improvement in their GERD-HRQL score off PPI at six months compared to baseline. Hence the study met its primary endpoint with the required 95% confidence level.
The reduction in the median score for the Brand X device of 23.0 units (from 29.0 to 6.0) represents a 79.3% improvement. This value is almost identical to the published result for the pivotal trial of the predicate device (79.2%). Therefore, the effectiveness of the Brand X system in successfully treating chronic symptoms of GERD is similar to the effectiveness reported for the predicate device.
The median value of the percent of time pH < 4.0 decreased from an initial value of 8.3% at baseline to 6.75%. Therefore, the study met its secondary endpoint related to the acid exposure test. A comparison to results reported in the literature revealed that the change in the median values of the Brand X device showed a decrease of 19%, while the predicate showed a decrease of 18%. Hence, the Brand X results in reducing the exposure to gastric acids are similar to those reported for the predicate system.
Safety
The study reported nine patients with a total of nine serious adverse events (SAEs). Four events were considered mild in intensity, involving pain and fever. Three events were classified as moderate in intensity, involving pneumothorax, pneumomediastinum, and pneumoperitoneum (all resolved spontaneously). Two events were considered severe in intensity: one involved esophageal perforation (required drainage) and another had suicidal thoughts (non-device/procedure related).
Six of the SAEs were considered related to the device: one definitely (esophageal perforation) and the others possibly. Three events were considered not related to the device. The median time from procedure to SAE was 1.5 days for events related to the device. None of the patients with SAEs required any operation or re-operation. Adverse events reported that occurred in greater than the 5% level were postoperative pain or discomfort in 33% of patients, postoperative nausea in approximately 10%, and sore throat in 21%. The adverse events were mild or insignificant in most cases.
The SAEs and overall safety profile were similar to the predicate device for which two perforations and one bleeding event were reported. The number of AEs was similar to those reported for the predicate device. Three cases of fever were reported in the current study (for 72 patients), similar to the 3 cases of fever reported for the predicate. There were 23 cases of chest pain (23/72 = 32%) vs. 17% reported for the predicate; abdominal pain was recorded for 5% of the patients in the current study vs. 44% of the patients for the predicate. Sore throat was reported for 15 patients (15/72 = 21%) vs. 15% for the predicate.
Summary
Based on the clinical performance as documented in the pivotal clinical study, the Brand X system was found to have a safety and effectiveness profile that is similar to the predicate device.
VIII. CONCLUSIONS
Since the predicate device was cleared based in part on the results of clinical studies, and since the comparison of bench testing to clinical outcomes is still not well understood for this type of device, clinical testing was required to support substantial equivalence. The non-clinical data support the safety of the device and the hardware and software verification and validation demonstrate that the Brand X device should perform as intended in the specified use conditions. The clinical data demonstrate that the Brand X device performs comparably to the predicate device that is currently marketed for the same intended use.
Appendix D. Glossary of Significant Terminology 🔗
The following terms are defined for purposes of this guidance:
Classification regulation – The classification regulations are Agency-defined categories of medical devices based on intended use and technology. Each device classification regulation defines the class (i.e., Class I, II, or III) for the device category which in turn determines the regulatory requirements. Device classification regulations are codified by rule or order in 21 CFR Parts 862-892.
Indications for use – The disease or condition the device will diagnose, treat, prevent, cure or mitigate, including a description of the patient population for which the device is intended.
Intended use – The general purpose of the device or its function. The intended use of a device encompasses the indications for use.
Multiple Predicate Devices – Two or more predicate devices that have been provided to support an SE determination. If using multiple predicate devices to demonstrate substantial equivalence, each predicate device must have the same intended use as the new device, and any different technological characteristics between the new device and the predicate devices must not raise different questions of safety and effectiveness.
Performance Data – Performance data can be any data, including non-clinical (e.g., data from engineering testing, such as fatigue, wear, corrosion, etc., biocompatibility, functional animal studies, cadaver, etc.) and/or clinical, that are provided to support the substantial equivalence of a device that is intended to be marketed.
Predicate Device – A legally marketed device (as defined in 21 CFR 807.92(a)(3)) to which a new device may be compared for a determination regarding substantial equivalence because the devices have the same intended use and the same technological characteristics or different technological characteristics that do not raise different questions of safety and effectiveness.
Primary Predicate Device – A predicate device with indications for use and technological characteristics that are most similar to the new device. The primary predicate should be identified within a 510(k) submission.
Reference Device – A legally marketed device that is intended to provide scientific and/or technical information (e.g., test methodology) to help address the safety and effectiveness of a new technological characteristic. Reference devices are not predicate devices and may only be used after Decision Point 4 on the 510(k) Decision-Making Flowchart.
Split Predicate – Using one legally marketed device for intended use and a different legally marketed device for technological characteristics to demonstrate substantial equivalence. The use of a “split predicate” is inconsistent with the 510(k) regulatory standard.
Footnotes 🔗
-
General controls apply to all classes of medical devices and provide FDA with the means of regulating devices to assure their safety and effectiveness. General controls include but are not limited to provisions that relate to establishment registration and device listing; premarket notification, although most class I devices are exempt by regulation from this requirement; prohibitions against adulteration and misbranding; records and reports; and good manufacturing practices. Section 513(a)(1)(A) of the FD&C Act (21 U.S.C. § 360c(a)(1)(A)). ↩
-
The original definition of a class II device in the Medical Device Amendments of 1976 (Pub. L. 94-295) identified performance standards rather than special controls as the mechanism by which FDA could establish reasonable assurance of safety and effectiveness. The Safe Medical Devices Act of 1990 (Pub. L. 101-629) added “special controls,” which can include the promulgation of performance standards as well as postmarket surveillance, patient registries, development and dissemination of guidelines (including guidelines for the submission of clinical data in premarket notification submissions), and other appropriate actions as FDA deems necessary to provide such assurance. Section 513(a)(1)(B) of the FD&C Act (21 U.S.C. § 360c(a)(1)(B)). ↩
-
Certain types of devices classified into class III that were in commercial distribution in the United States before May 28, 1976, and those determined to be substantially equivalent to such devices, may be cleared through the 510(k) process until FDA issues an administrative order requiring them to go through the premarket approval process. Section 515(b)(1) of the FD&C Act (21 U.S.C. § 360e(b)(1)). Prior to the enactment of the Food and Drug Administration Safety and Innovation Act (FDASIA) (Pub. L. 112-144) on July 9, 2012, FDA had to publish regulations to require such devices to go through the premarket approval process. Section 608(b) of FDASIA (126 Stat. 1056) changed the process from rulemaking to administrative order. ↩
-
For the purpose of this guidance document, a “new device” means a device within the meaning of section 201(h) of the FD&C Act that is not legally marketed. It can be either a completely new device or a modification of a legally marketed device that would require a new 510(k). ↩
-
By contrast, an unclassified devices, as defined in FDA’s Guidance for Industry and Food and Drug Administration Staff, “Medical Device Classification Product Codes” (http://www.fda.gov/MedicalDevices/DeviceRegulationandGuidance/GuidanceDocuments/ucm285317.htm), is a pre-amendments device for which a classification regulation has not been promulgated. Unclassified devices require submission of a 510(k) premarket notification to FDA. A not-classified device is a post-amendments device for which the Agency has not yet reviewed a marketing application or for which the Agency has not made a final decision on such a marketing application. A pre-amendments device is a device that was on the market prior to the enactment of the Medical Device Amendments to the FD&C Act on May 28, 1976. ↩
-
If FDA has established special controls applicable to the device type, the 510(k) would need to adequately address the issues covered by the special controls for the device to be classified into Class II. See Section 513(a)(1)(B) of the FD&C Act (21 U.S.C. § 360c(a)(1)(B)). ↩
-
See CDRH Preliminary Internal Evaluations – Volume I: 510(k) Working Group Preliminary Report and Recommendations (http://www.fda.gov/downloads/AboutFDA/CentersOffices/CDRH/CDRHReports/UCM220784.pdf). See also CDRH Preliminary Internal Evaluations – Volume II: Task Force on the Utilization of Science in Regulatory Decision Making Preliminary Report and Recommendations (http://www.fda.gov/downloads/AboutFDA/CentersOffices/CDRH/CDRHReports/UCM220783.pdf). See also 510(k) and Science Report Recommendations: Summary and Overview of Comments and Next Steps (http://www.fda.gov/downloads/AboutFDA/CentersOffices/CDRH/CDRHReports/UCM239449.pdf). ↩
-
Under 21 CFR 807.92(a)(3), a legally marketed device to which a new device may be compared for a determination regarding substantial equivalence is a device that was legally marketed prior to May 28, 1976, or a device which has been reclassified from class III to class II or I, or a device which has been found to be substantially equivalent through the 510(k) premarket notification process. ↩
-
Under section 513(a)(2) of the FD&C Act, the safety and effectiveness of a device are to be determined:
(A) with respect to the persons for whose use the device is represented or intended,
(B) with respect to the conditions of use prescribed, recommended, or suggested in the labeling of the device, and
(C) weighing any probable benefit to health from the use of the device against any probable risk of injury or illness from such use. ↩
-
Alternatives to PMAs include submission of a Product Development Protocol (PDP) (Section 515(f) of the FD&C Act) or a Humanitarian Device Exemption (HDE) (Section 520(m)(2) of the FD&C Act and 21 CFR 814.104). An HDE may be an appropriate option if the device has been determined by the Office of Orphan Products Development to be eligible for an HDE through a Humanitarian Use Device (HUD) designation (21 CFR 814.100 and 814.102). Unlike other regulatory submissions, given the limited patient population, an HDE only has to demonstrate a reasonable assurance of safety and probable benefit (Section 520(m)(2) of the FD&C Act and 21 CFR 814.104). For more information on HUD designations, please see FDA’s guidance “Humanitarian Use Device (HUD) Designations” (http://www.fda.gov/downloads/RegulatoryInformation/Guidances/UCM336515.pdf). ↩
-
The passage of FDASIA (Pub. L. 112-144) on July 9, 2012, included the Medical Device User Fee Amendments of 2012 (MDUFA III), Title II of FDASIA (126 Stat. 1002), which reauthorized the device user fee program for another five years. The MDUFA III Commitment Letter (http://www.fda.gov/downloads/MedicalDevices/NewsEvents/WorkshopsConferences/UCM295454.pdf) outlines changes to the review timeframes and/or expected interactions for many premarket submissions. ↩
-
See Preamendment Status (http://www.fda.gov/MedicalDevices/DeviceRegulationandGuidance/ComplianceActivities/ucm072746.htm). ↩
-
See 510(k) and Science Report Recommendations: Summary and Overview of Comments and Next Steps. (http://www.fda.gov/downloads/AboutFDA/CentersOffices/CDRH/CDRHReports/UCM239449.pdf). ↩
-
It is important to note that if multiple predicates are used to support the same intended use, any different technological characteristics between the new device and the cited predicate devices must not raise different questions of safety and effectiveness. Section 513(i) of the FD&C Act (21 U.S.C. § 360c(i)). ↩
-
The answer at Decision Point 2 may possibly be “no” if the predicate device is uncoated. Introducing a coated arthroplasty device into an anatomical location which previously only had non-coated devices would likely create a new intended use due to the different fixation methods. ↩
-
The applicability of the scientific methodology used to characterize certain aspects of a legally marketed device will depend upon the specific scenario. In this example, it is determined that the duration of contact, which affects the biocompatibility testing, and the mechanical testing conducted to fully characterize the coating on the hip implant are directly relevant and informative for the same coating applied to the knee implant. However, if the manufacturer wanted to rely on the scientific methodology for a coating used in a different type of implant (e.g., cardiovascular), it may not be appropriate to exercise this approach. ↩
-
Section 607 of FDASIA (Pub. L. 112-144; 126 Stat. 1054), which was enacted on July 9, 2012, amended section 513(f)(2) of the FD&C Act by providing the option of directly submitting a request for De Novo classification without the need for an NSE determination. A manufacturer who intends to submit a direct De Novo request is encouraged to engage in dialogue with FDA through the pre-submission process to obtain additional feedback related to whether a valid predicate exists for the new device and appropriate performance data that will be necessary to support a reasonable assurance of safety and effectiveness. For more information on the pre-submission process, see FDA’s Guidance for Industry and FDA Staff, “Requests for Feedback on Medical Device Submissions: The Pre-Submission Program and Meetings with Food and Drug Administration Staff” (http://www.fda.gov/downloads/MedicalDevices/DeviceRegulationandGuidance/GuidanceDocuments/UCM311176.pdf). ↩
-
Although devices recently cleared under the 510(k) program are often selected as the predicate device to which substantial equivalence is claimed, any legally marketed Class II or Class I device may be used as a predicate device. However, section 513(i)(2) of the FD&C Act provides that a predicate device may not have been removed from the market at the initiative of the Commissioner of Food and Drugs or been determined to be misbranded or adulterated by a judicial order. See also 21 CFR 807.100. ↩
-
This guidance is not intended to supplant either of the following guidance documents: “Determination of Intended Use for 510(k) Devices; Guidance for CDRH Staff (Update to K98-1)” (http://www.fda.gov/MedicalDevices/DeviceRegulationandGuidance/GuidanceDocuments/ucm082162.htm) or “General/Specific Intended Use” (http://www.fda.gov/MedicalDevices/DeviceRegulationandGuidance/GuidanceDocuments/ucm073944.htm). ↩
-
Refer to “Initial Results of 510(k) Audit: Analysis of Not Substantially Equivalent (NSE) Determinations” (http://www.fda.gov/AboutFDA/CentersOffices/OfficeofMedicalProductsandTobacco/CDRH/CDRHReports/ucm259173.htm). ↩
-
We have a long-standing policy of applying the definition of indications for use in the PMA regulation at 21 CFR 814.20(b)(3)(i) in the same way in the 510(k) context. ↩
-
For purposes of Section IV.D, the term “new” in describing indications for use refers to an indication that is new or differs from that of the predicate device. ↩
-
See 21 CFR 807.92(a)(5); see also FDA guidance “General/Specific Intended Use” (http://www.fda.gov/MedicalDevices/DeviceRegulationandGuidance/GuidanceDocuments/ucm073944.htm) which implements section 513(i)(1)(F) of the FD&C Act. 1 ↩
-
Section 513(i)(1)(A) of the FD&C Act and 21 CFR 807.100(b)(2). “Different technological characteristics” are defined as “significant change in the materials, design, energy source, or other features of the device from those of the predicate device.” Section 513(i)(1)(B) of the FD&C Act and 21 CFR 807.100(b)(2)(ii)(A). ↩
-
Refer to “Initial Results of 510(k) Audit: Analysis of Not Substantially Equivalent (NSE) Determinations” (http://www.fda.gov/AboutFDA/CentersOffices/OfficeofMedicalProductsandTobacco/CDRH/CDRHReports/ucm259173.htm). ↩
-
FDA’s regulations require manufacturers to include in their 510(k)s “[a] description of the device that is the subject of the premarket notification submission, such as might be found in the labeling or promotional material for the device, including an explanation of how the device functions, the scientific concepts that form the basis for the device, and the significant physical and performance characteristics of the device, such as device design, material used, and physical properties.” 21 CFR 807.92(a)(4); see also 21 CFR 807.87(f). ↩
-
The original Flowchart from the K86-3 Guidance included a decision point related to whether or not “descriptive characteristics” were precise enough to ensure equivalence. However, the term “descriptive characteristics” does not appear in the statute or regulations. The 510(k) Decision-Making Flowchart described in Appendix A specifically addresses this to reflect the statute more closely and minimize confusion. ↩
-
For additional considerations, refer to the FDA guidance, “Use of International Standard ISO-10993, 'Biological Evaluation of Medical Devices Part 1: Evaluation and Testing' (Replaces #G87-1 #8294) (blue book memo)” (http://www.fda.gov/MedicalDevices/DeviceRegulationandGuidance/GuidanceDocuments/ucm080735.htm). See also Draft Guidance for Industry and FDA Staff, “Use of International Standard ISO-10993, ‘Biological Evaluation of Medical Devices Part 1: Evaluation and Testing’” (http://www.fda.gov/downloads/MedicalDevices/DeviceRegulationandGuidance/GuidanceDocuments/UCM348890.pdf). FDA’s draft guidance represents the Agency’s proposed approach on this topic. ↩
-
We encourage manufacturers to determine whether there is an applicable device-specific guidance or special controls for the device type as provided in a special controls document or classification regulation. ↩
-
Manufacturers should also determine whether there is an applicable device-specific guidance or special controls for the device type as provided in a special controls document or classification regulation. ↩
-
FDA follows the “least burdensome” provisions. See Final Guidance for FDA and Industry, “The Least Burdensome Provisions of the FDA Modernization Act of 1997: Concept and Principles” (http://www.fda.gov/MedicalDevices/DeviceRegulationandGuidance/GuidanceDocuments/ucm085994.htm). ↩
-
The applicability of GLPs to non-clinical studies in a 510(k) submission is also mentioned in FDA’s Guidance “Refuse to Accept Policy for 510(k)s” in the Performance Data – General section of the checklist (http://www.fda.gov/downloads/MedicalDevices/DeviceRegulationandGuidance/GuidanceDocuments/ucm315014.pdf). ↩
-
21 CFR 860.7(c)(2): Valid scientific evidence is evidence from well-controlled investigations, partially controlled studies, studies and objective trials without matched controls, well-documented case histories conducted by qualified experts, and reports of significant human experience with a marketed device, from which it can fairly and responsibly be concluded by qualified experts that there is reasonable assurance of the safety and effectiveness of a device under its conditions of use. The evidence required may vary according to the characteristics of the device, its conditions of use, the existence and adequacy of warnings and other restrictions, and the extent of experience with its use. Isolated case reports, random experience, reports lacking sufficient details to permit scientific evaluation, and unsubstantiated opinions are not regarded as valid scientific evidence to show safety or effectiveness. Such information may be considered, however, in identifying a device the safety and effectiveness of which is questionable. ↩
-
In the U.S., clinical studies/investigations (see 21 CFR 812.3(h)) involving one or more human subjects to determine the safety or effectiveness of a device must be conducted in accordance with the Investigational Device Exemptions (IDE) regulations, 21 CFR Part 812, as applicable. In addition, such studies/investigations must comply with the regulations governing institutional review boards (21 CFR Part 56), informed consent (21 CFR Part 50), and financial disclosure (21 CFR Part 54). See also Guidance for Clinical Investigators, Industry, and FDA Staff, “Financial Disclosure by Clinical Investigators” (http://www.fda.gov/downloads/RegulatoryInformation/Guidances/UCM341008.pdf). Studies conducted outside the U.S. generally do not have to comply with the IDE regulations, but FDA recommends that such studies be conducted in accordance with good clinical practice. For information on good clinical practice, see 78 FR 12664 (Feb. 25, 2013). ↩
-
The acceptability or level of data necessary to support an SE determination is product specific and therefore, not discussed in this guidance. Manufacturers should determine whether there is an applicable device-specific guidance or special controls for the device type as provided in a special controls document or classification regulation as these sources may provide further information about performance data that may be necessary in a 510(k) submission. ↩
-
See SOP: Decision Authority for Additional or Changed Data Needs for Premarket Submissions (http://www.fda.gov/AboutFDA/CentersOffices/OfficeofMedicalProductsandTobacco/CDRH/CDRHReports/ucm279288.htm). ↩
-
As specified in 21 CFR 807.87(h), a 510(k) Statement as described in 21 CFR 807.93 may be provided in lieu of a 510(k) Summary. However, in order to facilitate transparency, FDA encourages all submitters to utilize the 510(k) Summary option. ↩
-
See Guidance for Industry and FDA Staff, “Bundling Multiple Devices or Multiple Indications in a Single Submission” (http://www.fda.gov/downloads/MedicalDevices/DeviceRegulationandGuidance/GuidanceDocuments/ucm089732.pdf). ↩
-
On July 9, 2012, section 605 of FDASIA (Pub. L. 112-144) added section 518A to the FD&C Act, which directs FDA to establish a program to routinely and systematically assess information regarding device recalls, and to use that information to proactively identify strategies for mitigating health risks presented by defective or unsafe devices. FDA believes that one way to carry out this directive is to provide greater transparency on recalled devices. Identifying whether a predicate was recalled is optional, but doing so would help the Agency achieve this FDASIA directive. ↩
