Innolitics helps design, develop, document, and validate software for SaMD and SiMD medical devices.
Whether you’re looking for support on a special project or a full-service medical software partner who can get you through the FDA, we can help.
All We Do Is Medical-Device Software
Innolitics has developed software for 70+ medical devices since
2012.
Cybersecurity
Our cybersecurity services include threat modeling, security risk
management, SBOMs, and more.
Medical-Device Standards
Our team has deep knowledge of DICOM, HL7, and other medical-device
standards.
Compliant Software Development
Our medical device software development services comply with ISO 13485
and IEC 62304.
FDA Compliance
We can write all software and cybersecurity documentation for FDA
submissions, with six FDA regulatory consultants on staff.
AI/ML
Our team includes four AI/ML and image-processing experts, including
three PhDs and two MDs.
SaMD and SiMD Expertise
Medtech OS, including its checklists, templates, and tooling, is built
from experience supporting FDA clearance for more than 50 SaMD and SiMD
products.
Every great partnership starts with a conversation. Fill out the form below for a discovery call, and an Innolitics team member will contact you soon.
I learned more from an hour-long Innolitics sales call than an entire paid engagement from other consultants.
Richard Clark
Executive in Residence at Washington University
Innolitics: Your Medical Device Software Development Partner
Innolitics is proud to partner with a diverse array of clients across the medical software industry, from visionary startups to industry-leading Fortune 500 companies. Typical clients include:
SaMD & SiMD Development: Building the Future of Healthcare
Our expertise spans a broad range of medical-device software applications, both Software as a Medical Device (SaMD) and Software in a Medical Device (SiMD) products.
We specialize in creating solutions that are not only innovative but also compliant and secure, ensuring they meet the highest industry standards.
We have experience developing a variety of medical device applications, including:
Medical Software Development Backed By Clinical Expertise
Our engineering work is guided by a clinical perspective. One of our founders is an MD and we have a broad network of clinicians we leverage. We have experience in the following clinical areas:
Radiology
Digital Health
Radiation Oncology
InVitro Diagnostics (IVDs)
Dental
Cardiology
Pre-Clinical
Ear Nose and Throat
Neurology
Gastroenterology and Urology
FDA Software Consulting for Medical Devices
We engage with our clients in three different ways to support medical device software at every stage of development, through FDA clearance and beyond.
Project-Based
We help you complete software development projects with a defined
scope. For example:
Custom Web-Based GUIs
Medical Imaging Software
SMART on FHIR apps
Adding DICOM or HL7 Integrations to existing products
Independent Software V&V for submissions
Refactoring Existing Software
Training New AI/ML Models
Image Processing Algorithm Development
Translating Research Code into Compliant Code
Developing Companion Apps
This engagement model works well when the scope of the project is
understood. We will work with you to define a scope, cost-estimate, and
timeline estimate for the project.
We provide experienced medical-device software
engineers to augment your in-house team.
Our engineers are trained on the FDA regulations and guidance and can
work within your QMS.
We provide engineers at either half-time or full-time and at varying
levels of experience. We can also increase and decrease staffing levels
as needed.
This engagement model is appropriate if you have poorly scoped work
or need ongoing support. Often, after completing a project-based
engagement, our clients request to retain the engineers they work with
in an on-going basis.
Regulatory Compliance for Regulated Medical Software Development
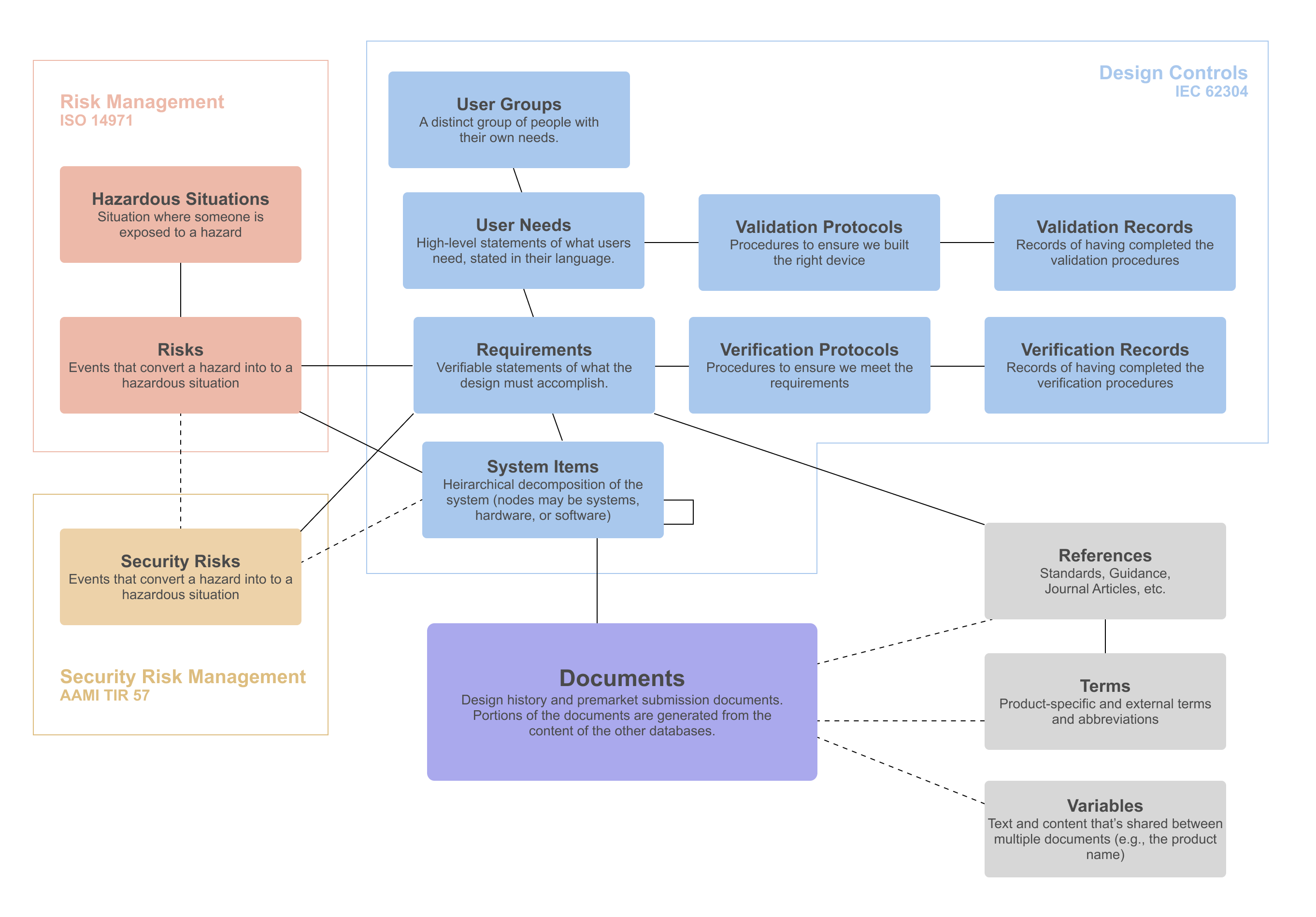
Our ISO 13485 compliant quality management system and development process adheres to the highest standards of quality and compliance, including IEC 62304 for medical device software, ISO 14971 for risk management, and AAMI TIR 57 for cybersecurity. Whether working within our quality management system or integrating with yours, we guarantee excellence and compliance at every stage.
Our Medical Device Software Technologies
Full-stack web-development, including TypeScript,
React, Sass and Django
Mobile app development (iOS and Android)
AI/ML model development, training, testing, and
deployment
Cloud deployments on Amazon Web Services (AWS), Microsoft Azure, and
Google Cloud
Native application development with Qt
Image and signal processing
Especially deep strengths in Python, TypeScript, Rust, C++, C, and
MATLAB
Embedded and real-time systems
Linux, Windows, and macOS
Cybersecurity for Medical Software Development
Our cybersecurity team can help you develop secure software that will
keep patients safe and your and your customer’s data secure.
Our Secure Product Development Framework (SPDF) incorporates:
Threat modeling
Security Risk Management (following AAMI TIR
57)
Security by Design
Software Bill of Materials (SBOM) generation
Cybersecurity Testing
Post-Market Vulnerability Monitoring
We can help you navigate navigate buyer’s IT concerns. We also help
with HIPAA. This includes providing copies of the MDS2 form and ensuring
your device makes it easy for your customers to comply with the HIPAA’s
security
rule.
Innolitics has deep expertise in image processing, signal processing
and AI/ML model development. Our algorithms have been deployed in
several FDA cleared applications and we can build your algorithm as part
of a new device or to augment an existing device. Our competencies
include:
Many medical device startups wisely keep their functionality to a
minimum for their first premarket submission. Once you're on the market,
customers often ask you to integrate with EMR Systems, Lab Information
Systems, and PACS.
We are DICOM experts. In fact, over 10,000 engineers and radiologists
visit our DICOM Standard
Browser each month, and it is often the top result in DICOM-related
Google searches.
Our medical software development services have helped SaMD and SiMD solutions get to market ahead of schedule and under budget.
I needed a software development partner to write the software, train the AI, and get FDA clearance. An investor once told me that it would take me $5 million and 5 years to get to where we are now. Innolitics got me here 3 years ahead of schedule and $4 million dollars under budget.
Dr. Andrew Smith, MD PhD
Co-Founder at AI Metrics &
Chair of Diagnostic Imaging at St. Jude Children’s Research Hospital
Innolitics’s engineers seamlessly integrate with our existing team. They’ve consistently produced high quality work while helping us solve technical problems in the medical imaging space. In particular, they spearheaded development of a large module that extends our existing product suite. This module has since been cleared by the FDA and is live at hospitals around the world. Their work on the module involved software development, researching various image processing algorithms, analysis of these algorithms against real-world data, and writing detailed technical documentation of the system that justified its approach to the FDA.
Nathan Childress, PhD, DABR
Associated VP at Varian Medical Systems
We needed a team that would get it done right the first time and independently. As a physician, I was delighted to work with another physician engineer on the team that was able to implement complex clinical workflows with very little input from me or my team. Innolitics delivered ahead of schedule and exceeded expectations. Every step of the way, Innolitics demonstrated why they’re leaders in medical software development.
Dr. Mark Rosenberg, DO, MBA, FACEP, FAAHPM, FACHT
Co-Founder and Chairman of Retrieve Medical
Innolitics is an incredible partner and consistently surpasses our expectations. They have an extremely agile team, adapting to our needs across back-end and front-end tasks seamlessly. When we needed support around ISO 62304 compliance for FDA requirements, they jumped right in and provided us compliant documentation. They also assisted us as we developed a regulatory strategy around FDA Cybersecurity and HIPAA Compliance. The Innolitics team is efficient, fair, and highly ethical. They are an absolute pleasure to work with.
Ryan Shelton, PhD
CEO and Co-Founder of PhotoniCare
We commissioned Innolitics to build a dashboard that integrated with our radiation-oncology EMR. Their team’s clinical experience greatly simplified communication and their perceptive questions and suggestions helped us arrive at a better set of requirements for the project. The final result was clean, easy for our doctors to use, and has already improved the efficiency of our workflow.
Jonas Fontenot, PhD
President and CEO of Mary Bird Perkins Cancer Center
This article discusses engineering and regulatory factors to consider when deciding to rewrite or reuse existing prototype code. It should be useful to researchers or companies who are repurposing existing software into a medical-device.
The FDA says developing your design inputs is “the single most important design control activity,” yet writing good design inputs is difficult. This article presents Innolitics’ answers questions our clients frequently ask us about design inputs and analyzes a number of poorly written example requirements.
Medical-device software release frequency is a common question, particularly from software engineers familiar with agile development. The answer hinges on two key factors: whether the changes require regulatory submissions and the ability to produce all necessary design change documentation quickly.
End-to-end SaMD projects can vary quite a bit depending on several
factors:
Whether the core algorithms or models have been proven
The size and complexity of the user interface
The number of platforms that are needed
The risk-classification of the device (higher-risk devices require
more documentation and testing)
The performance or hardware constraints
The number of integrations with external systems
And many other factors.
Historically, full end-to-end development of new SaMD projects has
ranged from $250k through multiple millions.
Many of our project-based engagements are much smaller.
What are your hourly rates?
Please reach out to use and we can provide you a list of our hourly
rates for the following roles:
Partner
Director
Senior Regulatory Consultant
Regulatory Consultant
Staff Software Engineer
Senior Software Engineer
Software Engineer
Data Annotation Associate
Do you ever work on a fixed-price basis?
No. We do not work on a fixed-price basis. For our project-based
engagements and end-to-end engagements we provide detailed cost and
timeline estimates. We refine and periodically update these estimates
throughout the project.
Can you provide examples of past projects similar to our product?
Yes! Please review our
case studies for a sampling of our past projects. If you don’t see
anything relevant, please reach out as only a small number of our
projects have case studies.
How do you handle project management and communication during the development process?
For staff-augmentation projects we assume you will be managing our
resources along with the rest of your team.
For our project-based and end-to-end engagements we provide project
management support. Here are a few of the highlights:
We set up preferred communication channels during kickoff (often
Slack or Microsoft Teams)
We follow an agile process with one- or two-week sprints
During these meetings we demo our work and plan the next sprint
We provide periodic budget and timeline estimate updates
What experience does your team have with FDA regulatory submissions for medical devices?
We have deep experience with FDA regulatory submissions. We have
three full-time regulatory consultants on our team. Collectively we have
cleared over forty-five 510(k) submissions and several De Novos. Our
engineers are also trained on writing the software and cybersecurity
design history documentation required for FDA submissions.
In short, we are among the most experienced FDA regulatory experts in
the world when it comes to medical device software.
Does your team do firmware development?
Several engineers on our team have experience with firmware
development, however, we typically are not the best fit for
firmware-heavy projects. We excel on Software in a Medical Device (SiMD)
projects that involve a lot of application-level coding, such as web,
mobile, or desktop UIs, cloud deployments, image processing, etc.
Can your team help design our user interface?
Yes. Typically we will develop an initial set of UI Mockups in Figma
and will then have a Medical Device UI/UX designers polish and further
refine the slides.
What if your engineers don’t know a core technology that we use?
Good engineers can usually become proficient using new databases,
frameworks, or even programming languages pretty quickly. Tools like
GitHub Co-Pilot and ChatGPT have made it learning new technologies even
easier.
If this is a major concern, we occasionally will cover the cost for
training engineers on new technologies.
Can your engineers work within our QMS?
Yes! We can work within your QMS or within ours QMS. We are happy to
sign a quality agreement to coordinate quality activities. We can also
train our engineers within your QMS. This often makes sense for Staff
Augmentation engagements.
Can you help us set up a Quality Management System (QMS)?
Yes. We can get you set up on our Medtech OS platform. This is the
platform we use for our QMS. You can read more about
Medtech OS here.
Can you run our QMS?
Sorry, although we’re often asked to do this, we do not currently
help companies run their QMS, however, we can provide guidance and
support. We do have several partners whom we can recommend who can help
you run your QMS.
What is your usage of AI in medical device software development?
While we incorporate AI at various points in our delivery process, we
do not treat AI-assisted work any differently from work produced without
it — we take ownership of all deliverables. AI amplifies our existing
value proposition of speed and certainty.
Agentic AI tools like Claude Code and Codex are changing the day-to-day
experience of software engineering. We see this as an opportunity, not a
threat. We are a growing, collaborative team that is actively learning
how to use these tools to produce robust, traceable, high-quality
systems where correctness, safety, and documentation matter.
As an engineer here, you will not only write production code, but also
develop expertise in medical device regulations, quality systems,
verification and validation, and the realities of FDA-facing
software
Get Medical Device Software Support From Innolitics
Whether you need to augment your medical software staff or need an end-to-end software partner, Innolitics can help you clear hurdles and get through the FDA.