About this Transcript 🔗
This document is a transcript of an official FDA (or IMDRF) guidance document. We transcribe the official PDFs into HTML so that we can share links to particular sections of the guidance when communicating internally and with our clients. We do our best to be accurate and have a thorough review process, but occasionally mistakes slip through. If you notice a typo, please email a screenshot of it to Mihajlo at mgrcic@innolitics.com so we can fix it.
Preamble 🔗
Document issued on September 28, 2022.
Document originally issued on February 9, 2015.\
For questions about this document regarding CDRH-regulated devices, contact the Digital Health Center of Excellence by email at digitalhealth@fda.hhs.gov. For questions about this document regarding CBER-regulated devices, contact the Office of Communication, Outreach, and Development (OCOD) at 1-800-835-4709 or 240-402-8010, or by email at ocod@fda.hhs.gov.
Contains non-binding guidance.
I. Introduction 🔗
The Food and Drug Administration (FDA) recognizes that the progression to digital health offers the potential for better, more efficient patient care and improved health outcomes. To achieve this goal requires that many medical devices be interoperable with other types of medical devices and with various types of health information technology. The foundation for such inter-communication is hardware and software, typically referred to as medical device data systems (MDDS) that transfer, store, convert formats, or display medical device data and results. This guidance provides FDA’s current thinking for MDDS, as well as medical image storage devices and medical image communications devices, to provide clarity and predictability for manufacturers on these devices.
In general, FDA’s guidance documents do not establish legally enforceable responsibilities. Instead, guidances describe the Agency’s current thinking on a topic and should be viewed only as recommendations, unless specific regulatory or statutory requirements are cited. The use of the word should in Agency guidances means that something is suggested or recommended, but not required.
II. Background 🔗
On February 15, 2011, the FDA issued a regulation down-classifying MDDS from Class III (high-risk) to Class I (low-risk) (“MDDS regulation”).1 Since down-classifying MDDS, FDA has gained additional experience with these types of technologies, and has determined that these devices pose a low risk to the public. On February 9, 2015, FDA issued a guidance document to inform manufacturers, distributors, and other entities that the Agency does not intend to enforce compliance with the regulatory controls that apply to MDDS, medical image storage devices, and medical image communications devices, due to the low risk they pose to patients and the importance they play in advancing digital health.
Since the issuance of the guidance document in 2015, section 3060(a) of the 21st Century Cures Act (Cures Act) amended section 520 of the Federal Food, Drug, and Cosmetic Act (FD&C Act) on December 13, 2016, removing certain software functions from the definition of device in section 201(h) of the FD&C Act. Specifically, pursuant to section 520(o)(1)(D) of the FD&C Act, software functions that are solely intended to transfer, store, convert formats, or display medical device data or medical imaging data, unless the software function is intended to interpret or analyze clinical laboratory test or other device data, results, and findings, are not devices and are not subject to FDA laws and regulations applicable to devices. On September 27, 2019, this guidance was previously revised through a minor update in accordance with the Cures Act, with the changes described in the guidance “Changes to Existing Medical Software Policies Resulting from Section 3060 of the 21st Century Cures Act.”2 The policy described in this guidance document is also consistent with the Agency’s updated guidance, also revised through a minor update on September 27, 2019, entitled “Policy for Device Software Functions and Mobile Medical Applications,” originally issued on February 9, 2015, with the title “Mobile Medical Applications.”3
This guidance is being now revised through a minor update to be consistent with the final rule “Medical Devices; Medical Device Classification Regulations To Conform to Medical Software Provisions in the 21st Century Cures Act” (86 FR 20278, April 19, 2021).4 This rule amended certain medical device classification regulations to reflect changes in the FD&C Act made by the Cures Act, as described above. FDA amended these medical device classification regulations so that our regulations conform to the medical software provisions in the Cures Act. This latest revision also includes minor changes to address comments submitted to the docket. This guidance reflects FDA’s current thinking on the definitions of MDDS, medical image storage devices, and medical image communications devices.
III. Definitions 🔗
The following terms are used for the purposes of this guidance:
Non-Device-MDDS: Software functions that are solely intended to transfer, store, convert formats, or display medical device data and results.
Device-MDDS: Hardware functions that are solely intended to transfer, store, convert formats, or display medical device data and results.
IV. Policy for Non-Device-MDDS and Device-MDDS 🔗
A. Policy for Non-Device-MDDS 🔗
Software functions that meet the definition of Non-Device-MDDS, medical image storage devices, or medical image communications devices are not devices under section 201(h) of the FD&C Act. As such, software functions that are solely intended to transfer, store, convert formats, or display medical device data and results, including medical images, waveforms, signals, or other clinical information, are not devices and thus are not subject to FDA regulatory requirements applicable to devices. However, software functions that analyze or interpret medical device data in addition to transferring, storing, converting formats, or displaying clinical laboratory test or other device data and results remain subject to FDA’s regulatory oversight unless they meet the criteria outlined in section 520(o)(1)(E) of the FD&C Act.
FDA further clarifies the Non-Device-MDDS policy through the following examples:
A Non-Device-MDDS is a software function solely intended to provide one or more of the following uses, without controlling or altering the functions or parameters of any connected medical devices, which may or may not be intended for active patient monitoring:5
- The electronic transfer or exchange of medical device data. For example, this would include software that collects output from a ventilator about a patient’s carbon dioxide level and transmits the information to a central patient data repository.
- The electronic storage and retrieval of medical device data. For example, software that stores historical blood pressure information for later review by a health care provider.
- The electronic conversion of medical device data from one format to another in accordance with a preset specification. For example, software that converts digital data generated by a pulse oximeter into a digital format that can be printed.
- The electronic display of medical device data, which may include certain secondary or remote displays to medical devices that solely display data and results. For example, software that displays a previously stored electrocardiogram for a particular patient.
Non-Device-MDDS do not modify the data, do not control the functions or parameters of any connected medical device, and do not analyze or interpret data. Software functions intended to generate alarms or alerts or prioritize patient-related information on multi-patient displays, which are typically used for active patient monitoring, are considered device software functions because these functions involve analysis or interpretation of laboratory test or other device data and results. As noted above, a Non-Device-MDDS may include software functions that transfer, store, convert formats, or display medical device data that may or may not be intended for active patient monitoring. Software functions that are device functions intended for active patient monitoring to enable immediate awareness for potential clinical intervention include the following characteristics:
- The clinical context requires a timely response (e.g., in-hospital patient monitoring).
- The clinical condition (disease or diagnosis) requires a timely response (e.g., a monitor that is intended to detect life-threatening arrhythmias, such as ventricular fibrillation, or a device used to actively monitor diabetes for time-sensitive intervention).
Examples of devices that provide active patient monitoring include:
- A nurse telemetry station that functions as a secondary alarm system that receives and displays information from a bedside hospital monitor in an intensive care unit (ICU) to enable immediate awareness for potential clinical intervention.
- A device that receives and/or displays information, alarms, or alerts from a monitoring device in a home setting and is intended to alert a caregiver to take an immediate clinical action.
Examples of software functions that transfer, store, convert formats, or display medical device data and are Non-Device-MDDS:
- An application that transmits a child’s temperature to a parent/guardian while the child is in the nurse/health room of a school.
- An application that facilitates the remote display of information from a blood glucose meter, where the user of the meter can independently review their glucose and glucose levels, and which is not intended to be used for taking immediate clinical action. In these cases, remotely displaying information such as the most recent blood glucose value or time-lapse between blood glucose measurements is not considered active patient monitoring.
- Any assemblage or arrangement of network components that includes specialized software expressly created for a purpose consistent with the definition of Non-Device-MDDS.
- Custom software that is written by entities other than the original medical device manufacturer (e.g., hospitals, health care providers, third party vendors) that directly connects to a medical device solely to obtain medical device information.
- Modified portions of software that are part of an Information Technology (IT) infrastructure created and/or modified (writing and compiling software) for specific Non-Device-MDDS functionality.
B. Policy for Device-MDDS 🔗
Hardware functions that are intended to transfer, store, convert formats, or display medical device data and results remain devices under section 201(h) of the FD&C Act. FDA does not intend to enforce the requirements under the FD&C Act for hardware functions that are considered to be Device-MDDS, medical image storage devices, or medical image communications devices, provided that the hardware function is limited to assisting the following software functions: electronic transfer, storage, conversion of formats, or display of medical device data. Device-MDDS do not modify the data and do not control the functions or parameters of any connected medical device. These hardware functions may include the following regulations:
- MDDS subject to 21 CFR 880.6310,6
- Medical image storage devices subject to 21 CFR 2010,7 and
- Medical image communications devices subject to 21 CFR 2020.8
This means that for hardware functions that meet the definitions in the regulations listed above, FDA does not intend to enforce compliance with the regulatory controls, including registration and listing, premarket review, postmarket reporting, and quality system regulation for manufacturers of these types of devices.
Each Device-MDDS regulation listed above contains an exemption from premarket notification; however, limitations to this exemption identified under 21 CFR 880.9 and 21 CFR 892.9 require a premarket notification in the listed circumstances. Even when exceeding these limitations, FDA does not intend to enforce compliance with the regulatory controls for hardware functions that meet the definitions identified by the above regulations. For example, to the extent that these limitations apply, FDA does not intend to enforce compliance with regulatory controls for a Device-MDDS that is an in vitro device that is intended for assessing the risk of cardiovascular diseases (21 CFR 880.9(c)(4)) or for use in diabetes management (21 CFR 880.9(c)(5)).
Specialized medical display hardware devices for digital mammography, radiology, pathology, and ophthalmology (see, for example, 21 CFR 892.2050) and other specialized medical display hardware integral to the safe and effective use of a medical device hardware product (such as integral 3D displays in robotic surgery systems and displays built into ICU bedside monitors) have not been considered MDDS, medical image storage devices, or medical image communications devices. Such medical display hardware devices and other specialized medical display hardware integral to a medical device are not excluded from the device definition by the Cures Act and are not considered to be Device-MDDS.
In some cases, software functions that transfer, store, convert formats, or display medical device data and results are utilized on hardware that is not intended by the hardware manufacturer for a device function under section 201(h) of the FD&C Act. For example, general-purpose hardware IT infrastructure intended for data transfer (e.g., network router), data storage (e.g., network attached storage (NAS)), conversion of data (e.g., PDF software), and display of data (computer monitor) are not device functions. Such products do not meet the definition of a device in section 201(h) of the FD&C Act for either the software or hardware function and are therefore not regulated as devices.
C. Multiple Function Device Products that contain MDDS 🔗
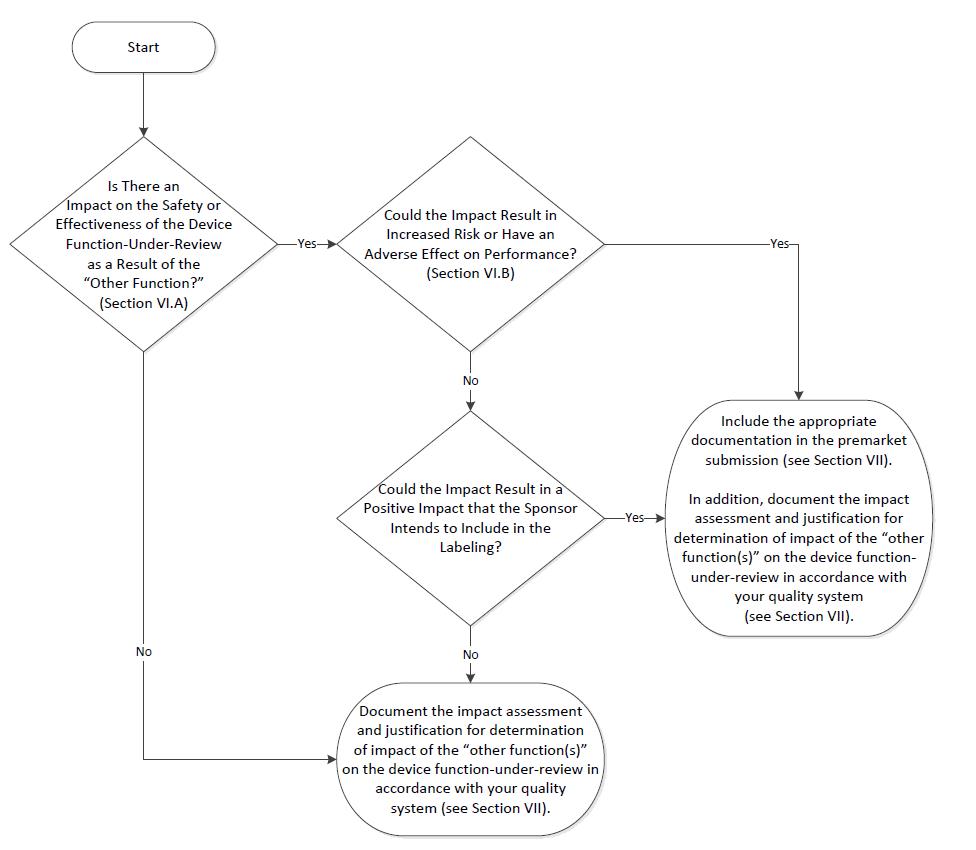
Consistent with section 520(o)(2) of the FD&C Act, which describes the regulation of a product with multiple functions, including at least one device function and at least one software function that is not a device, FDA does not regulate the Non-Device-MDDS functions contained in a multiple function device product. If a multiple function device product contains Device-MDDS functions, FDA does not, at this time and based on our current understanding of the risks of these devices, intend to enforce the requirements under the FD&C Act. However, FDA may assess the impact that such Non-Device-MDDS and Device-MDDS functions have on the safety and effectiveness of the device function(s) in the multiple function device product. FDA’s recommendations on the regulation of such products with multiple functions is provided in the guidance “Multiple Function Device Products: Policy and Considerations.”9
Footnotes 🔗
-
See the final rule “Medical Devices; Medical Device Data Systems” (76 FR 8637) at https://www.federalregister.gov/documents/2011/02/15/2011-3321/medical-devices-medical-device-data-systems. ↩
-
Available at https://www.fda.gov/regulatory-information/search-fda-guidance-documents/changes-existing-\ medical-software-policies-resulting-section-3060-21st-century-cures-act. ↩
-
Available at https://www.fda.gov/regulatory-information/search-fda-guidance-documents/policy-device-softwarefunctions-and-mobile-medical-applications. ↩
-
See 86 FR 20278 at https://www.federalregister.gov/documents/2021/04/19/2021-07860/medical-devices-medicaldevice-classification-regulations-to-conform-to-medical-software-provisions. ↩
-
As noted in the preamble of the MDDS regulation, the word “active” represents “any device that is intended to be relied upon in deciding to take immediate clinical action” (76 FR 8637 at 8644). FDA further noted that there are existing classifications for patient monitoring devices (See, e.g., 21 CFR Part 880, Subpart C (general hospital and personal use monitoring devices); 21 CFR Part 868, Subpart C (anesthesiology monitoring devices); 21 CFR Part 884, Subpart C (obstetrical and gynecological monitoring devices); and 21 CFR Part 870, Subpart C (cardiovascular monitoring devices)). ↩
-
Available at https://www.ecfr.gov/current/title-21/chapter-I/subchapter-H/part-880/subpart-G/section-880.6310. ↩
-
Available at https://www.ecfr.gov/current/title-21/chapter-I/subchapter-H/part-892/subpart-B/section-892.2010. ↩
-
Available at https://www.ecfr.gov/current/title-21/chapter-I/subchapter-H/part-892/subpart-B/section-892.2020. ↩
-
Available at https://www.fda.gov/regulatory-information/search-fda-guidance-documents/multiple-functiondevice-products-policy-and-considerations. ↩