
Innolitics introduction 🔗
Innolitics provides US FDA regulatory consulting to startups and established medical-device companies. We’re experts with medical-device software, cybersecurity, and AI/ML. See our services and solutions pages for more details.
We have practicing software engineers on our team, so unlike many regulatory firms, we speak both “software” and “regulatory”. We can guide your team through the process of writing software validation and cybersecurity documentation and we can even accelerate the process and write much of the documentation for you (see our Fast 510(k) Solution).
About this Transcript 🔗
This document is a transcript of an official FDA (or IMDRF) guidance document. We transcribe the official PDFs into HTML so that we can share links to particular sections of the guidance when communicating internally and with our clients. We do our best to be accurate and have a thorough review process, but occasionally mistakes slip through. If you notice a typo, please email a screenshot of it to Mihajlo at mgrcic@innolitics.com so we can fix it.
Preamble 🔗
Draft document issued on December 19, 2023.
For questions about this document regarding CDRH-regulated devices, contact the Office of Clinical Evidence and Analysis at CDRHClinicalEvidence@fda.hhs.gov. For questions about this document regarding CBER-regulated devices, contact the Office of Communication, Outreach, and Development (OCOD) at 1-800-835-4709 or 240-402-8010, or by email at ocod@fda.hhs.gov.
Contains non-binding guidance.
I. Introduction 🔗
FDA is issuing this draft guidance to clarify how FDA evaluates real-world data to determine whether they are of sufficient quality for generating real-world evidence that can be used in FDA regulatory decision-making for medical devices. This draft guidance also provides expanded recommendations to sponsors considering using real-world evidence to support a regulatory submission for medical devices.1
Real-world data (RWD) are data relating to patient health status and/or the delivery of health care routinely collected from a variety of sources.
Examples of RWD sources include data derived from electronic health records (EHRs),2 medical claims data, data from product and disease registries, and data gathered from other sources (such as digital health technologies) that can inform on health status. RWD sources can be used as data collection and analysis infrastructure to support many types of study designs, including, but not limited to, randomized and non-randomized controlled trials; single-arm studies with or without comparison to an objective performance criterion, performance goal, or extended control; observational studies; and hybrid designs which combine elements of multiple study designs.
Real-world evidence (RWE) is the clinical evidence regarding the usage, and potential benefits or risks, of a medical product derived from analysis of RWD.
This draft guidance includes factors that FDA considers important to demonstrate whether the RWD are fit-for-purpose for a particular regulatory decision relating to medical devices, as well as FDA’s recommendations on how FDA intends to assess these factors. When finalized, the recommendations and considerations in this draft guidance will apply regardless of the RWD source and encompass processes for conducting studies to generate RWE. A fit-for-purpose assessment should evaluate both the relevance and reliability of the RWD, discussed in more detail in Section V. FDA recognizes that there may be other approaches to address the considerations identified in this document. We encourage sponsors to discuss their approach with FDA, especially if the approach diverges from the recommendations in this draft guidance, when finalized.3
FDA recognizes and anticipates that the Agency and industry may need up to 60 days to perform activities to operationalize the recommendations within the final guidance. At this time, the Agency anticipates that, for regulatory submissions that will be currently pending with FDA after publication of the final guidance, as well as those submissions received within 60 days following publication of the final guidance, FDA generally would not anticipate that sponsors will be ready to include the newly recommended information outlined in the final guidance in their submission. FDA, however, would intend to review any such information if submitted at any time.
In general, FDA’s guidance documents do not establish legally enforceable responsibilities. Instead, guidances describe the Agency’s current thinking on a topic and should be viewed only as recommendations, unless specific regulatory or statutory requirements are cited. The use of the word should in Agency guidances means that something is suggested or recommended, but not required.
II. Background 🔗
To protect and promote public health, FDA needs to understand and evaluate the available evidence related to regulated products.For medical devices, common sources of available evidence include non-clinical and clinical studies4 provided to FDA by a device manufacturer or sponsor of a device premarket or postmarket submission. FDA recognizes that a wealth of clinical data in the form of RWD are routinely collected in the course of clinical practice during the treatment and management of patients. Although these data typically have different quality controls compared to data collected within a traditional clinical study, under certain circumstances RWD may be used to generate RWE to help inform or augment FDA’s understanding of the benefit-risk profile of devices at various points in their life cycle. Per 21 CFR 860.7(c)(1), “[a]lthough [a] manufacturer may submit any form of evidence to the Food and Drug Administration in an attempt to substantiate the safety and effectiveness of a device, the agency relies upon only valid scientific evidence to determine whether there is reasonable assurance that the device is safe and effective.” RWE derived from relevant and reliable RWD may constitute valid scientific evidence,5 depending on the study question, regulatory decision, data source(s), and design and analysis of the specific dataset derived from RWD source(s), and thus may be used to support regulatory decisions. FDA intends to use the considerations described in this draft guidance, when finalized, to evaluate whether RWD are relevant and reliable to support regulatory decision-making, including potentially generating valid scientific evidence. The use of RWE for specific regulatory purposes will include assessment of the overall relevance and reliability of the RWD used to generate the RWE.
When appropriate, use of RWD may provide an efficient means of generating the necessary clinical evidence to support regulatory decisions. Information specific to the clinical performance of a device can be generated through a number of methodological and operational approaches. In general, traditional clinical studies tend to be narrow in scope but allow for more control of sources of error and bias. In comparison, studies that leverage RWD may be able to evaluate broader questions but are subject to sources of bias which may be more difficult to control.
Clinical evidence can be generated from studies using RWD, alone or in combination with data from more traditional clinical studies. Using appropriate design and methodologies, sponsors can leverage the strengths of these approaches while minimizing potential weaknesses.
RWD that includes patient experience data6 may provide new insights into the performance of a device. In addition, RWD may foster inclusion of target populations that are otherwise underrepresented in clinical studies. Similarly, leveraging RWD may allow for studies of a longer period of time than would be practical in a traditional clinical study and so may allow for data to be gathered on longer term outcomes. Clinical evidence generated from fit-for-purpose RWD informs device benefit-risk profiles assessment from a real-world environment, allows evaluation of outcomes which may not be feasible in traditional clinical studies, and better aligns with device innovation cycles to inform future device modifications and new technology development. Finally, RWD may include information from broader clinical experiences than is usually represented in traditional clinical studies. RWE is an important factor for understanding and regulating medical devices, and therefore, FDA encourages the medical community to learn more from routine clinical care to help support safety and effectiveness of medical devices. Use of relevant and reliable RWD to generate RWE can benefit stakeholders throughout the ecosystem, including but not limited to, patients, health care providers, manufacturers, and FDA.
Additionally, in some cases, a traditional clinical study may be impractical or excessively challenging to conduct. Ethical issues regarding treatment assignment, and other similar challenges, may present themselves when developing and attempting to execute such a study. Analyses of RWD, using appropriate methods, may in some cases provide similar information with comparable or even superior characteristics to information collected and analyzed through a traditional clinical study. Under the right conditions, RWE may be suitable to support the marketing authorization of a new device or the expansion of the indications for use of devices that are already on the market. Aggregation of RWD (e.g., in a medical device registry7) may prove useful as a postmarket control suitable for providing ongoing device safety surveillance and additional evidence for effectiveness. FDA has long applied postmarket controls as a way to reduce premarket data collection, while still ensuring that the statutory standard of reasonable assurance of safety and effectiveness is met.8 FDA believes that applying postmarket controls to reduce premarket data collection, when appropriate, can help improve patient access to safe and effective medical devices.9 Many of the considerations and best practices for generating RWE are derived from the same principles that govern generation of clinical evidence from traditional clinical studies, which are generally referred to as good clinical practice (GCP). Additionally, as with all clinical evidence FDA evaluates, FDA’s assessment of RWE evaluated in support of a particular regulatory decision will be included as part of the totality of information available to FDA. Further, as with all types of clinical data, FDA recognizes there may be uncertainty of the benefits and risks of a device that remain after completion of a study using RWD. Some of these aspects are similar to those also present in more traditional forms of clinical data, but some are unique to RWD.
Assessment of the relevance and reliability of the RWD, as outlined in this draft guidance, can 133 identify uncertainty that should be considered during the benefit-risk determinations10 for the 134 device for a given regulatory purpose.
In 2017, FDA issued the guidance document, Use of Real-World Evidence to Support Regulatory Decision-Making for Medical Devices,10 in which we described the relevance and reliability factors of RWD that FDA assesses to determine if RWD are sufficient for generating RWE. Subsequently, on December 29, 2022, the Food and Drug Omnibus Reform Act of 2022 ("FDORA") was signed into law as part of the Consolidated Appropriations Act, 2023, Pub. L. No. 117-328. Section 3629 of FDORA "Facilitating the Use of Real World Evidence" directs FDA to issue or revise existing guidance on considerations for the use of RWD and RWE to support regulatory decision-making. FDA is issuing this draft guidance to propose revisions to the 2017 guidance, Use of Real-World Evidence to Support Regulatory Decision-Making for Medical Devices, to satisfy the requirement under section 3629(a)(2). This draft guidance also fulfills a commitment in Section V.F. of the Medical Device User Fee Amendments Performance Goals and Procedures, Fiscal Years 2023 Through 2027 (MDUFA V).11 This draft guidance is intended to provide expanded and updated recommendations to industry and FDA staff for conducting an assessment of relevance and reliability to demonstrate that RWD may be fit-for-purpose to generate clinical evidence for regulatory decision-making. This includes recommendations to provide clarity on least burdensome general expectations related to demonstrating that RWD is fit-for-purpose for premarket regulatory purposes.
III. Scope 🔗
This draft guidance is applicable for the use of RWE to support regulatory submissions for medical devices.12
The topics covered within this draft guidance are framed specifically for the use of RWD and RWE in regulatory submissions for medical devices (e.g., Investigational Device Exemption (IDE), premarket notification under section 510(k) of the FD&C Act, Premarket Approval Application (PMA), Humanitarian Device Exemption (HDE), De Novo classification request, post-approval study, postmarket surveillance under Section 522 of the FD&C Act (522 submissions), Clinical Laboratory Improvement Amendments (CLIA) Waiver by Applications (CW), Dual De Novo/510(k) and CLIA Waiver by Application Submissions (Duals)). The considerations included in this draft guidance may be applicable to supporting uses of RWD across the medical device total product life cycle (TPLC).
This draft guidance does not address the use of non-clinical data, adverse event reports, secondary use of clinical study data, or systematic literature reviews. Nor does it address all possible study design/conduct or analytical methodologies. While it does describe the factors that FDA considers when evaluating relevance and reliability of RWD, it does not provide a specific set of criteria or other scoring tools for determining the suitability of any specific RWD source for generating RWE for a particular regulatory decision.
This guidance, when finalized, should not be construed to alter or change in any way the existing evidentiary standards applicable to FDA’s regulatory decision-making. Rather, this guidance describes the circumstances under which clinical evidence generated from RWD may be used to support a variety of FDA decisions based on the existing evidentiary standards. While FDA encourages the use of relevant and reliable data to generate clinical evidence, including RWE, this draft guidance neither mandates use of RWD and RWE nor restricts other means of providing evidence to support regulatory decision-making. This draft guidance does not affect any federal, state, or local laws or regulations, or foreign laws or regulations that may be applicable to the use or collection of RWD, or that provide protections for human subjects (including informed consent requirements) or patient privacy. When finalized, this guidance should be used to complement, but not supersede, other device-specific and GCP and guidance documents.
IV. Regulatory Context in Which Use of RWE May be Appropriate 🔗
A. General considerations for the use of RWE 🔗
In general, FDA considers the use of RWD to be fit-for-purpose to support generation of clinical evidence for regulatory decision-making for medical devices when we conclude that the RWD used to generate the RWE are relevant to and reliable for informing or supporting a particular regulatory decision. It is important to understand the strengths and limitations of the underlying RWD and how these qualities impact their relevance and reliability. Similarly, the context of the specific regulatory decision for which the RWE is being proposed is central to FDA’s evaluation.
FDA recognizes that RWE can be generated from a variety of RWD sources that are primarily intended for another purpose. For example, administrative claims data13 are typically collected for purposes of billing or payment for medical care. Disease-specific RWD sources may be useful for tracking progression or outcomes of specific rare or poorly understood diseases. Treatment-specific RWD sources may have several purposes, including assessment and tracking of overall outcomes, providing assessment of hospital operations, informing performance improvement initiatives, or providing risk prediction and benchmarking data for specific therapies. The suitability of the RWD source may be determined by the factors outlined in Section V. and the availability of sufficient data to address the study question of interest.
FDA does not endorse one type of RWD over another. Sponsors should select the appropriate RWD sources based on their suitability to address the specific study questions. Data sources that may be considered RWD sources include the following:14
- Registries;15
- EHRs;
- Administrative claims data;
- Patient-generated data16 created, reported, or gathered by patients including in-home use settings (e.g., data from digital health technologies (DHTs)17 such as wearables);
- Device-generated data (e.g., implantable devices, physiological monitoring devices);
- Public health surveillance data (e.g., COVID-1918 case surveillance);
- Clinically annotated biobanks; and
- Medical device data repositories (e.g., imaging, electrocardiography databases).
Some purposes for which use of RWD may potentially be applicable in a regulatory submission include the following:
- To generate hypotheses to be tested in a clinical study;
- As a historical control, an informative prior in a Bayesian analysis of a clinical trial,19 or as one source of data in a hierarchical model or a hybrid data synthesis;
- As a concurrent control group or as a mechanism for collecting data to support marketing authorization when a registry, EHR, claims data, or some other systematic data collection mechanism exists;
- As a mechanism for re-training artificial intelligence/machine learning-enabled medical devices;
- To generate evidence to identify, demonstrate, or support the clinical validity of a biomarker or clinical outcome assessment;
- To generate (primary) clinical evidence to support marketing authorization (e.g., HDE, PMA, 510(k) or De Novo request);
- To generate evidence directly by the subject device to provide new information on safety or effectiveness;
- To generate evidence to support a determination on whether the subject device meets the statutory criteria for a CLIA waiver20 (e.g., CW and Duals21);
- To generate evidence to support the interpretability of the primary clinical evidence (e.g., to demonstrate that the study population for an investigation conducted outside the United States (OUS) is representative of the US population, or to provide context for an adverse event observed in the clinical study);
- To generate evidence to support a petition for reclassification of a medical device under section 513(e) or (f)(3) of the FD&C Act;
- To generate evidence for expanding the labeling of a device to include additional indications for use or to update the labeling to include new information on safety and effectiveness;22
- To generate evidence for postmarket surveillance. Through ongoing surveillance, signals are at times identified that suggest there may be a safety issue with a medical device. RWE may be generated using RWD to refine these signals for purposes of informing appropriate corrective actions and communication;
- To conduct post-approval studies that are imposed as a condition of device approval or to potentially preclude the need for 522 submissions; and
- To provide postmarket data in lieu of some premarket data, consistent with FDA’s policy on balancing premarket and postmarket data collection.23
B. Application of Investigational Device Exemption (IDE) Requirements in 21 CFR 812 to the Collection of RWD 🔗
An approved IDE permits a device to be shipped lawfully for the purpose of conducting investigations of the device without complying with certain other requirements of the FD&C Act that would apply to devices in commercial distribution. The purpose of this investigational exemption, per 21 CFR 812.1, “is to encourage, to the extent consistent with the protection of public health and safety and with ethical standards, the discovery and development of useful devices intended for human use, and to that end to maintain optimum freedom for scientific investigators in their pursuit of this purpose.” As explained in 21 CFR Part 812, the IDE regulations apply to all clinical investigations of devices to determine safety and effectiveness, with certain limited exceptions.24 In many cases, an approved IDE is required before initiating a clinical investigation. An investigation is defined as “a clinical investigation or research involving one or more subjects to determine the safety or effectiveness of a device.”25
Whether the collection of RWD for a legally marketed device requires an IDE depends on the particular facts of the situation. Specifically, if the device is being used in the normal course of medical practice, an IDE would likely not be required. FDA recognizes that in clinical practice this could include use of a legally marketed device for uncleared or unapproved uses, where the device is being administered or prescribed under the authority of a health care practitioner within a legitimate practitioner-patient relationship. If data collection does not impact how the device is administered, and the administration is within the normal course of medical care, an IDE would likely not be required. For example, analyses of extant RWD (i.e., RWD already collected) involving the use in medical care of a device that was not within the cleared or approved indications for use would generally not be subject to IDE regulations. However, similar to traditional clinical studies, if data are being gathered to determine the safety and effectiveness of the device, and the process for gathering the data would influence treatment decisions, such administration would likely not be within the normal course of medical practice and an IDE may be required. For example, a study using a registry infrastructure designed to determine the safety and effectiveness of an approved device for a new intended use would likely be subject to IDE requirements if physicians are instructed to treat specific patients or otherwise administer the device in a prescribed way for purposes of data generation, or when certain follow-up activities are performed for the purpose of research.
Should a sponsor or Institutional Review Board (IRB) be unclear regarding the applicability of the IDE regulations to a particular RWD collection activity or use, the sponsor or IRB should contact FDA. If an IDE is determined to be required, FDA intends to work with the IDE sponsor to develop the least burdensome approach to facilitate the efficient generation of RWE. Note that regardless of the applicability of 21 CFR Part 812, FDA regulations at 21 CFR Part 56 (IRB review), Part 50 (Protection of Human Subjects) and Part 54 (Financial Disclosure) may apply to RWE generation activities, as may other federal, state, and local laws regarding human subject protections.
C. Application of RWD from devices authorized for emergency use under section 564 of the FD&C Act 🔗
Section 564 of the FD&C Act provides that FDA may, after the HHS Secretary has made a declaration of emergency or threat justifying authorization of emergency use (an “EUA declaration”), authorize the emergency use of an unapproved product26 or an unapproved use of an approved product for certain emergency circumstances.
The routine clinical use of a device authorized under an EUA, when used within the scope of its authorization, is not considered to be a clinical investigation (see section 564(k) of the FD&C Act and Section IV.B. for more information on the application of IDE requirements in 21 CFR Part 812 to the collection of RWD). Clinical data routinely collected from the use of a device authorized under an EUA may be considered RWD and may be used to support regulatory decision-making, if determined to be fit-for-purpose. Generally, the recommendations in this draft guidance may apply to RWD from devices authorized under an EUA. Additionally, Appendix B includes an example of RWD from a device authorized under an EUA that was used in a subsequent premarket submission.
Device use pursuant to EUAs may lead to additional sources and novel uses of RWD to support FDA decision-making. We encourage sponsors to consider the recommendations in this guidance for devices authorized under an EUA (e.g., devices authorized under an EUA during the COVID-19 pandemic).
V. Assessing Data Relevance and Reliability 🔗
To determine the potential suitability of RWD to generate RWE for regulatory decision-making, FDA assesses the relevance and reliability of the RWD source as well as the data elements, study design, and analytic components of the study. This section describes the elements that might be evaluated to determine if the data are fit-for-purpose. FDA recognizes that data, including RWD used to generate RWE, may have limitations. Sponsors should understand the strengths and limitations of generating evidence from RWD to address a specific study question and provide these limitations to FDA in their submission. If the RWD source appears relevant and reliable, then additional assessment of the study-specific derived dataset(s) may help demonstrate the RWD are fit-for-purpose to address the study question. This assessment may be used to determine whether the RWD source(s) and the proposed design and analysis can generate evidence that is sufficiently robust to be used for the given study question and regulatory purpose, i.e., whether the RWD are fit-for-purpose.
Whether data are sufficiently relevant and reliable for use will, in part, depend on the particular regulatory decision. FDA will evaluate the same factors to assess RWD across all data sources and regulatory decisions but will weigh each factor in accordance with the regulatory decision to be made. In cases where RWE is derived from multiple RWD sources, each RWD source will be evaluated individually and together in the aggregate to determine the relevance and reliability of the RWD.
The data should be accurate, as complete as possible, and of adequate data quality to credibly address the question at hand. Conducting a clinical investigation in accordance with GCP provides assurance that the data and results from the clinical investigation are credible and accurate and that the rights, safety, and well-being of subjects are protected.27 In traditional clinical studies, the best practices for incorporating GCP into the study design and execution are generally well established. However, because RWD are typically collected outside of a controlled research setting, additional precautions should be considered to ensure that the data are similarly “credible and accurate,” and that appropriate patient protections are in place. In order to determine whether data are “credible and accurate,” FDA assesses the relevance and reliability of the data.
Additionally, sponsors should ensure that RWD were collected using good data management practices and are sufficiently robust. Sponsors should also consider data related to various demographic characteristics (e.g., age, sex, race, and ethnicity)28 and other potentially relevant covariates, and whether the data are representative of the intended use population. The relevance and reliability factors listed below should be described to assess the RWD.
Studies using RWD should also be carefully designed to mitigate potential bias, and a study protocol and analysis plan should be created prior to analyzing RWD, regardless of whether the RWD are extant or if they are to be collected in the future. An existing RWD source may have some inherent sources of bias that could limit the relevance or reliability for drawing causal inferences between medical device exposures29 and outcomes.
To help ensure the relevance and reliability of the source data, FDA recommends sponsors consider the factors contained in this section. If considered, these factors should be referenced during study conduct, FDA inspection, or provided as additional information during FDA review of the applicable regulatory submission, as applicable. Appendix A sets forth the elements that FDA recommends sponsors document and have available for inspection, as well as recommended elements sponsors to include in the appropriate regulatory submission for FDA review.
A. Relevance 🔗
Relevance includes consideration of availability, timeliness, and generalizability of the RWD. When needed information is not available in one data source, sponsors may want to provide linkage of other data source(s). Important relevance factors that FDA will consider in determining whether RWD are suitable for generating RWE for regulatory use include the following:
(1) Data availability 🔗
The RWD should contain sufficient detail to capture the information needed to evaluate the question being addressed in the target population. Relevant considerations should include whether the RWD contains information on the following:
- Use of the device (e.g., the device identifier (DI)30 portion of the unique device identifier (UDI),31 other structured data, clinical notes) or other exposure in the study population;
- Outcome(s) of interest in the study population;
- Covariates that may impact the exposure or outcomes of interest (e.g., RWD source contains signs, symptoms, treatments, procedures, diagnoses, patient and family history, pre-existing conditions, labs, demographics, and results which may be used to construct covariates that are relevant to the study question); and
For example, the minimum set of data fields in a registry may be insufficient for a specific study question and additional data fields may be needed for the registry to be fit-for-purpose. The registry should retain information documenting the start or stop of collection during the study time frame for data fields related to the specific study question. - Longitudinality, including longevity (the length of time that data for an individual is captured within the RWD source) and continuity of care.
Information across the continuum of care32 (i.e., data observability) may aid the assessment of the likelihood that all exposures and outcomes of interest will be captured for regulatory decision-making.
For example, tertiary care hospitalization data may not have adequate data availability to study outcomes that are likely to be diagnosed in an emergency for all patients, because patients are likely to go to a nearby hospital in emergencies but may travel to another location for a specialty device procedure.
If the RWD source is insufficient on its own, the sponsor should determine whether supplemental data sources are available and sufficient to provide any missing information necessary to address the study question.
(2) Linkages 🔗
419 Sponsors should assess whether and how data from different sources can be obtained and integrated given the potential for heterogeneity in target population characteristics, clinical practices, and coding across data sources. A description of this assessment should be provided in the regulatory submission for FDA review.
Any linkages performed within and across RWD sources should use a predefined linkage methodology33 that is scientifically valid, protects the privacy of individuals whose data will be used, supports interoperability, and accounts for differences in coding and reporting across sources. The following considerations should be assessed by the sponsor:
- Adequacy of line-level linkages (i.e., that the same individuals are being matched), including pre-defined rules to check for logical consistency and value ranges to confirm that data were retrieved accurately from a linked data source; and
- Application of strategies to correct for redundant data, to resolve any inconsistencies, and assess the potential for missing data.
Because patients typically visit multiple health care sites, especially in geographically contiguous areas, the inclusion of de-identified data from many sites creates the possibility that there will be multiple records from different health care sites for a single individual. This can result in overcounts of a particular data measure. Alternatively, if some site records are not available, this can result in a collection of histories that reflect only a fraction of the patient’s total health care history.
(3) Timeliness 🔗
As with traditional clinical studies, the time between data collection and release for research should be reasonable and the RWD considered for the study should reflect the current clinical environment (e.g., RWD from before a major change in clinical practice may not be timely). Sponsors should consider changes in clinical practice and guidelines over time (e.g., criteria for disease diagnosis, cancer staging), characteristics of a condition (e.g., prevalent strain of a pathogen) and health status of the population. If data are being collected within the RWD source during the study time frame, then the sponsors should update the availability of the RWD in a timely manner and should define the reporting schedule in the regulatory submission for FDA review.
(4) Generalizability of data 🔗
Once a study question is defined, the specific study sample meeting inclusion and exclusion criteria should be (1) representative of the population in the RWD source eligible for use of the device within the specified indication and (2) generalizable to the target population with the condition of interest. If upon quantitative assessment, the study sample is shown to not be representative of a subset of the target population, then analyses should be conducted to evaluate generalizability of the study findings.
B. Reliability 🔗
Reliability includes consideration of accrual, quality, and integrity of RWD. Important reliability factors that FDA considers in determining whether the RWD are suitable for generating RWE for regulatory use include the following:
(1) Data Accrual 🔗
To ensure the reliability of the RWD source, data should be collected and processed in a consistent and methodical manner. The manner of collection may differ for newly developed RWD sources which are actively collecting data (e.g., data dictionary to provide a common definitional framework in a registry), using nationally or internationally recognized coding systems (e.g., International Classification of Diseases, Tenth Revision, Clinical Modification (ICD-10-CM), Logical Observation Identifiers, Names, and Codes (LOINC), UDI, Current Procedural Terminology (CPT) in EHR or Claims) or custom-designed structured data capture (e.g., data capture within a device), or using unstructured data capture (e.g., narrative portion of clinical notes). Any of these approaches may be able to demonstrate sufficient reliability to support regulatory decision-making. Factors FDA will consider in making this determination include:
- Adequacy of information and descriptors about data sources provided in the regulatory submission for FDA review, which should include information on:
- Data types;
- Health care settings/environment(s);
- Purpose of data collection;
- How data were obtained at point of data capture;
- How data are accessed by study team and sponsor;
- Any data transformations, including any modifications made for privacy protection;
- Full data dictionary or common data capture form, if applicable;
- Device information, including types of identifiers (e.g., DI) and indication for use;
- Completeness of fields that would typically be completed for all participants and needed for most study questions (e.g., age, sex, DI);
- Time frame (including common temporal framework for collection of data) and latency of the data (including the timeliness of data entry, transmission, and availability);
- Version control; and
- Key technical and privacy-related information. Sponsors should document routine migration of data between various sources over time (e.g., indicate the date and time of data transfers, linkages).
- Adequacy of information about data accrual methods and procedures provided in the regulatory submission for FDA review, which should include information on:
- Site collection procedures;
- Use of common data capture forms;
- Common definitional frameworks;
- Data cleaning and cross-referencing procedures;
- The sources and technical methods used for data element capture (e.g., chart abstraction, point of care entry, EHR integration, UDI capture, data records from the device, and linkage to administrative claims data); and
- Methods for data retrieval and processes to minimize missing data extraction, implausible values, and data quality checks in data captured at the point of care (e.g., during clinical practice for manual or automated health care data collection processes) to ensure accuracy and completeness of core data fields.
(2) Data Quality and Integrity 🔗
When considering RWD sources for regulatory purposes, sponsors should consider the methods and systems used to help ensure sufficient data quality, including any data quality assurance plans and procedures developed for the RWD source itself. Since evaluation of RWD sources may not always permit specific line-item source verification, important factors for sponsors to consider include:
- Quality control processes;
- Regardless of the original purpose for collecting the RWD, procedures for data collection and quality assurance should be put into place during the data source design and development stages to optimize the reliability, quality, and usefulness of the data, as appropriate. These procedures should be described in the regulatory submission for FDA review.
- Where appropriate, processes should include site and data monitoring, data quality audit programs, and evaluation of ongoing training programs for data collection.
- Records regarding the assessment of adherence to the RWD source’s established data quality assurance and quality control policies and procedures should be retained.
- Assessment of completeness, accuracy, and consistency across sites and over time;
- Data should be captured in a manner designed to minimize missingness. Missingness and out of range values should be assessed for each data element. The amount of missingness per participant (across data elements) should also be assessed. The impact of missingness should be considered and thresholds for unacceptable levels of missingness should be pre-determined. Additionally, quantitative assessment of the potential bias associated with high missingness should be performed and included in the interpretation of the study.
- Data should be reflective of the actual patient experience (e.g., interactions with health care, disease trajectory, outcomes) with the condition of interest.
- Consistency of data capture should be used across sites and over time.34 If any changes are needed (e.g., where diagnostic criteria, definitions, or clinical practice change over several years), then sponsors should document those changes and assess their impact on the study question and provide summary information in the regulatory submission for FDA review.
- Auditing rules, methods, and the mitigation strategies used to reduce errors should be documented.35
- Study sample size should be adequate to address the study question.
- If non-extant data are used (e.g., data for a newly marketed device will be captured in the future using the infrastructure of an existing data source), the sample size should be determined based upon adequate statistical power to detect a clinically meaningful difference.
- If extant RWD are used, adequate statistical power to detect a clinically meaningful difference should be determined based on the available sample size and should account for any sampling of participants from the data source.
- If there is inadequate statistical power based on the available sample size, sponsors should consider the use of multiple existing RWD sources to increase sample size.
- If the sample size could be expected to increase in the near
- future (e.g., device is new to market), sponsors should consider conducting “interim” analysis with extant data, monitoring uptake, and conducting final analysis when sufficient sample size is available.
- Sponsors should account for planned statistical analysis within the study size calculations (e.g., 1:1 matching of propensity scores in a study population where 10% of participants receive device would remove approximately 89% of participants with comparator from the analysis).
- Establishment and adherence to data collection, recording, and source verification procedures;
- As with all clinical evidence generation, data provenance and traceability are important. Sponsors should plan and document all aspects of data extraction, aggregation, curation, storage, and availability for research, as described below.
- Sponsors should ensure any automated electronic transmission of data fields to a repository (e.g., registry or data warehouse) occurs in a consistent and reproducible fashion.
- Adherence to source verification procedures and data collection and recording procedures should be documented for completeness and consistency.
- Data checks and procedures should be prespecified to help address identified errors (e.g., in coding or interpretation of the source documentation or transformation).
- Sponsors should describe the mitigations used to address audit findings, including data corrections.
- Sponsors should identify the source document(s) and first instance36 of data available to sponsor. Sponsors should generate data quality documentation from the first instance through RWD dataset(s) used to address the study question.
- If using a common data model, sponsors should ensure documentation of the transformation of data from the original source to the common data model is retained.
- Data audit trail, including assessment of discrepancies, should be included.
For extant data sources, the sponsor may have access to the initial capture of data (e.g., direct access to EHR data as obtained via data entry) or may only have access to a partially curated data source (e.g., administrative claims data, aggregated EHR). We recommend sponsors maintain information on the data audit trail from the first instance of data available to the sponsor through all aspects of data analysis. Further, sponsors should obtain as much information as possible about the audit trail from the data holder if the first instance is not at the point of data entry. - When the RWD source is not owned by the sponsor, the sponsor should attempt to obtain participant-level data for each participant. If not available, the sponsor should define the entity(ies) which do have access/permission for data entry, quality assurance, storage, aggregation or other linkage, and assessment of traceability from data entry to dataset, as applicable. Sponsors should consider the level of access which could be shared with FDA and the potential for third parties to provide participant-level data directly to FDA. The availability of data should be described in the regulatory submission for FDA review.
- As with all clinical evidence generation, FDA recommends that the sponsor have access to the RWD source from the first instance and to the RWD dataset used for the analyses throughout the regulatory decision-making process. FDA recognizes that some data sources will not allow sponsors to access the participant-level data. Although we do not discourage use of these data sources, FDA notes that uncertainty may arise if the sponsor does not have access to all of the necessary data.
- Adequate patient protections (e.g., methods to protect the privacy of individuals’ health data and adherence to applicable privacy and ethics standards) established in advance of executing the study protocol; and
- Prior demonstration of RWE generation from the data source.
- Sponsors should provide documentation (including fit-for-purpose assessment) of any previous use of the same RWD source for a similar target population and peer-reviewed literature of RWE generation from the data source.
VI. Considerations for Methodologies for Collection and Analysis of RWD to Generate RWE 🔗
A study using relevant and reliable RWD in a well-designed and rigorously analyzed manner may be less burdensome than a traditional clinical study. Just as traditional clinical studies should be carefully designed, studies using RWD also should undergo careful assessment before embarking on the study or during the analysis to assure that the data are fit-for-purpose. FDA recognizes that some regulatory decisions may not be adequately supported using RWE, for various reasons, and we therefore recommend that sponsors consider the methodologies described below to address factors that can impact interpretability of a study using RWD.
Scientifically sound clinical study planning in advance of statistically valid analyses is important regardless of whether a study uses a traditional clinical study approach, uses only RWD, or incorporates a hybrid design. FDA recommends that any study be informed by the needs of the study question and regulatory decision driving the evidence generation. Further, just like for traditional clinical studies and in addition to the study design and analysis considerations described in Sections VI.A. and VI.B., FDA recommends that a sponsor document their decisions and the associated rationale for the following:
- Whether to include randomization, concurrent, or historical controls;
- The choice of performance goals and objective performance criteria;
- Type I and type II error control;
- Data gathering or dependence on extant data;
- Bias mitigation strategies;
- Precision of outcome measures and other data elements, as applicable; and
- All other known factors pertinent to interpretation of the study results.
Although many of the considerations in this section for data collection and analysis are not novel in the context of clinical evidence generation, there may be unique aspects of these considerations for studies using RWD. Additionally, the information presented in this section is intended to augment, not replace, information in other FDA guidances on the design of clinical studies for regulatory decision-making. The information in this section is intended to clarify implementation of these concepts and practices when using RWD. In particular, the information below is intended to complement information in the FDA guidance, “Design Considerations for Pivotal Clinical Investigations for Medical Devices.”37
A. Methods for study designs using RWD 🔗
Generally, FDA does not endorse a specific type of study design for clinical studies, regardless of whether it is a traditional clinical study or uses RWD. As with all clinical evidence generation, choosing the appropriate design for studies using RWD depends on the study question, device, outcome, key covariates, and the specific study objectives or hypotheses. Additionally, sponsors should consider the regulatory purpose of the generated clinical evidence. FDA recognizes that multiple types of study designs may also be useful to generate RWE. These study designs may include:
- Single-arm studies with comparisons to external controls, in whole or part;
- Objective performance criteria or performance goals;
- Non-interventional studies (observational studies) (e.g., comparative cohort studies, case-control studies, self-controlled studies, and descriptive studies); and
- Randomized controlled trials using RWD to supplement one or more study arms.
Furthermore, FDA recognizes the utility of RWD in assessing device utilization, participant characteristics, natural history of disease or disease trajectory, treatment environment and treatment patterns, as well as background rates of outcomes.38
B. Defining study design elements 🔗
For studies using RWD, as with all clinical studies, after determining the overarching study design, the study time frame and collection of data elements should be defined, followed by a system to capture specific data elements (e.g., in a case report form). Additional data capture requirements may necessitate justification or adjudication, especially for study endpoints. FDA recommends clearly defining the individual data elements derived from the RWD source to develop study-specific RWD. Similarly, FDA recommends that sponsors show that the data elements, as defined and applied within the study design, are relevant, reliable, and fit for the regulatory purpose. For analysis of RWD and interpretation of RWE, sponsors should have a study design that describes the study time frame, the pre-defined set of data elements, and a systematic consideration that the proposed data elements are all necessary for inclusion and represent all the key data elements.
(1) Study time, relative to index date 🔗
In traditional clinical studies, a participant is often enrolled into the study, has a baseline visit, and then first uses the device or has a procedure on the “index date” (see Figure 1). After that, the participant is usually followed for a period of time until a final visit. Data elements are collected at each visit, although different information may be gathered at each visit, and additional data elements may be collected outside of clinical care (e.g., via a participant diary or wearable). The participant continues to be followed through a last study visit. If a visit or other data collection is missed, then the participant may be contacted, or additional questions may be asked at the next visit to gather key information.
Figure 1. Traditional clinical studies - study time frame relative to index date
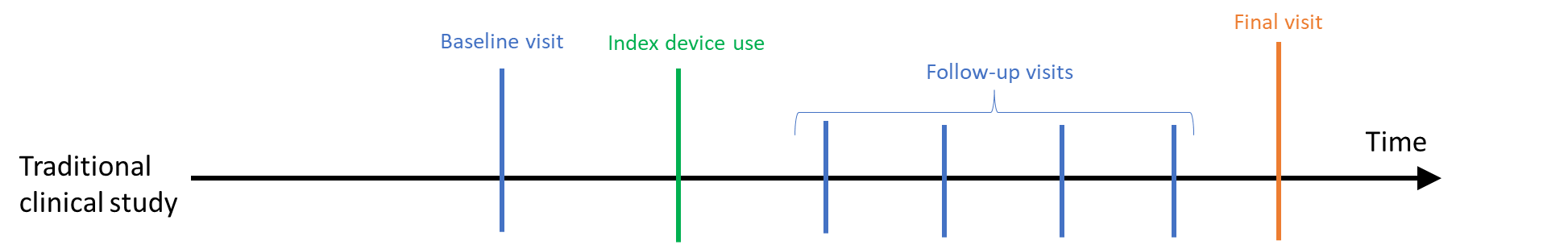
In clinical studies using RWD where data are collected after the study is designed, it is possible that a similar visit structure and data collection will be available (e.g., within a registry). However, follow-up visits may not occur on a set schedule or more patient-generated data may be collected. For extant data such as EHR or administrative claims, baseline and follow-up data are not collected on a set schedule; rather data collection coincides with clinical care over a period of time (see Figure 2). Participants may also enter or exit the source database as their life situation changes (e.g., move out of a geographic area or a change in health insurance). Thus, continuity of care is an embedded part of the study.
A visual depiction, such as that exemplified in Figure 2, may be helpful in identifying the timing of collection for each data element relative to the index date, which will help to identify potential bias. Data elements that may impact the initial device use should be collected before or at the time of initiation. Outcomes of device use occur after use of the device is initiated. Additionally, the index date for the use of the comparator to the device would occur at a similar point in the progression of disease. As with a traditional clinical study, discussion of these ideas with the study team or with FDA may be aided by the visual depiction of when each data element will be assessed.
Figure 2. Clinical study using RWD - study time frame relative to index date
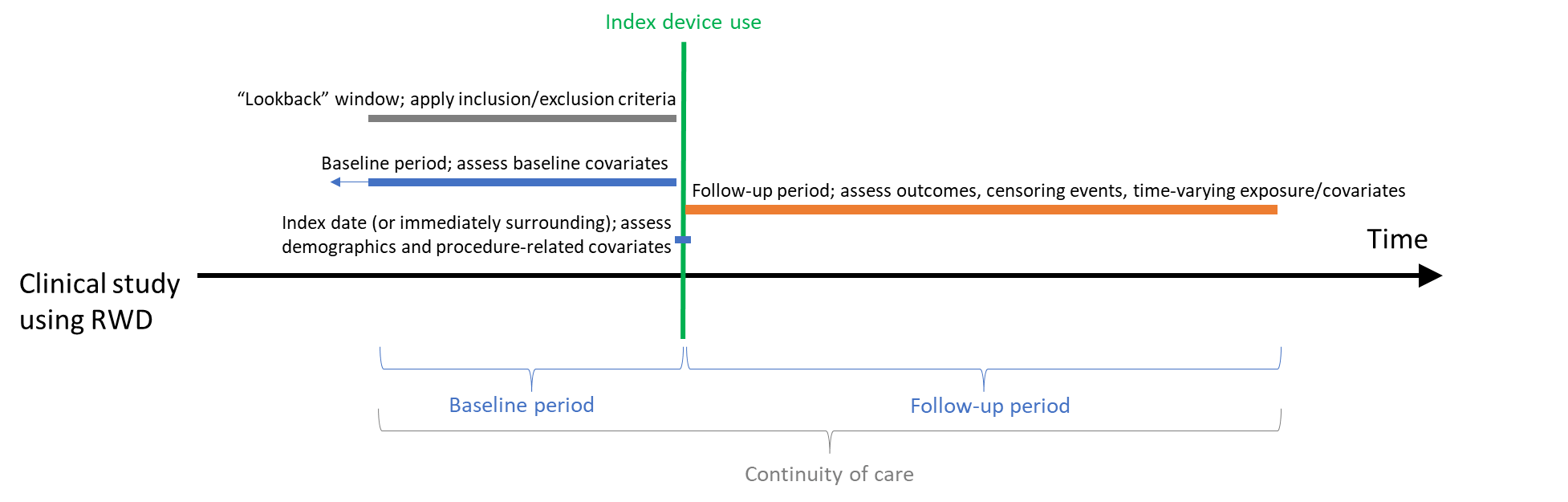
Follow-up in a study using RWD typically extends from the index date of device use until either the end of the pre-planned follow-up time or the last time identified within the RWD source. FDA considers the study end date to be the last date that participant follow-up could occur. This date is set on a day when data checks/audits can assure that the underlying data are of sufficient quality for use in research. Any data in the RWD source indicating that a participant had subsequent care is no longer included in the study (i.e., study participation is censored on this date). Thus, similar to the first site being ready for enrollment in traditional clinical studies, FDA expects that the study time frame will be defined to begin on the earliest date that the first data element could be collected and extend through the latest date that the last data element could be collected.
In addition to the study design elements discussed above, any change in the standard of care, availability of the device or other treatments, or other relevant factors (e.g., change in hospital care due to a public health emergency) should be included on the graphical depiction. This additional information may aid in systematically capturing these time-dependent data elements and provide support for their inclusion as covariates in analyses or consideration of sensitivity analysis (e.g., assessing whether a change in ICD-CM coding from a prior edition or a major change in clinical practice affects study results).
The calendar time allotted for the study should be long enough to adequately measure all data elements in a study – from the beginning of the baseline period through the end of the follow-up needed to assess the outcome(s) of interest – in a sample of participants large enough to provide adequate statistical power to detect the minimal clinically important difference in the primary outcome.
(2) Development of conceptual and operational definitions for the study population, device, comparator, outcome, and covariates 🔗
As with any clinical study, all data elements should be defined before the start of a study using RWD and should address the specific study question when valid and appropriate analytical methods are applied (i.e., the data are amenable to sound clinical and statistical analysis). A “conceptual definition” that describes the construct or feature of each data element in general or quantitative terms should be generated using a shorthand name or notation. This conceptual definition should reflect the current medical and scientific thinking regarding the variable of interest, such as: (1) clinical criteria to define a condition for population selection or as an outcome of interest or a covariate; or (2) measurement of the device or procedure to define an exposure of interest. For example, a conceptual definition might be “acute myocardial infarction (AMI)” or “AMI evidenced by increased troponin.” For a traditional clinical study, the sponsor defines the collection and timing of each data element, whether at a visit or between visits, and usually has the ability to contact the participant to limit missing data or to solicit additional information if a visit was missed. In a study using RWD, the data elements may be collected in a similar fashion (e.g., registry) or need to be defined from clinical care visits (e.g., EHR or administrative claims data) or some other algorithm (e.g., combining unstructured EHR and patient-reported data).
An “operational definition” describing all of the components needed to identify complete and accurate data elements from the data source should also be generated. While an operational definition would typically be generated in a case report form in a traditional clinical study, operational definitions in a study using RWD frequently include combining structured codes or unstructured notes (e.g., clinical notes) in an algorithm to identify presence of the data element. FDA considers the operational definition to include three components, as applicable:
- Time frame over which assessment occurs;
- Specific codes/component(s) assessed (e.g., via code lists); and
- Algorithm for combining the components (leading to positive identification or lack of identification). If machine learning is used to define criteria, sponsors should provide a full description of data management practices including the specifications of the model/algorithm (e.g., training, tuning, and testing), data collection and the data attributable to the proposed intended use population (e.g., with respect to race, ethnicity, disease severity, sex, age, socioeconomic characteristics), as well as verification and validation information to indicate that the machine learning approaches are fit-for-purpose for defining criteria.
The availability of different data types in studies using RWD may make it possible to establish operational definitions that are different from those typically used in traditional clinical studies. These definitions may or may not be appropriate in the context of the study question being addressed depending on the study question and regulatory purpose. It is important to consider whether the operational definition will capture the intended concept for each data element and FDA notes that small differences in the choice of operational definition in a specific data source (e.g., requiring two diagnoses rather than one diagnosis of AMI in the example above) may have a large impact on study results (e.g., considerably decrease the identification of the disease or condition under study). FDA considers minimizing misclassification to be a critical part of the process of defining an operational definition. FDA recommends reviewing previous studies using the RWD source, including published literature, and gathering expert opinion when developing operational definitions. For some data elements, a rationale for the operational definition based on previous studies or expert opinion may be sufficient. Some data elements may warrant more scrutiny to ensure that the interpretation of study results is not substantially impacted by their misclassification or missingness.
In some cases, it may be appropriate to conduct a validation study in which quantitative measurements of the operational definition are compared to a “ground truth” reference standard. This may result in updating the operational definition to ensure that these critical data elements are accurately identified. When conducting a validation study, a protocol should be developed before initiating the data collection and analysis specific to the validation. The protocol generally contains the plan to compare the operational definition in the RWD (e.g., administrative claims data) with the “ground truth” in the reference standard (e.g., validating that administrative billing diagnosis accurately represents a point-of-care diagnosis by comparing an operational definition in administrative claims against an EHR) and prespecification of the acceptance criterion for each validation measure (e.g., sensitivity, specificity, or positive and negative predictive values) which is of interest.
As with traditional clinical studies, in choosing existing operational definitions or developing new ones, sponsors should maximize identification of those who have the condition and to minimize incorrectly identifying those without the condition as having it (i.e., minimize misclassification). Sponsors can check for misclassification, for example, by generating a table of the proportions of participants within the study who are at each level of each data element and performing a qualitative comparison to what is known from previous literature or expert opinion about that data element in the target population. Further exploration is recommended for data elements that are not aligned with expectation. This same exercise may help identify any under-recording or missingness of data elements within the study.
In some RWD sources, the data elements that would be preferred for traditional clinical studies may not be available to the sponsor. However, a proxy for this missing information could be developed based on the information that is collected in the RWD source. Proxies can be developed for a wide range of uses, including identifying study participants (i.e., applying inclusion/exclusion criteria) as well as certain study endpoints. It may be possible to use the proxy, but sponsors should determine the suitability of a proxy by considering whether the proxy is clinically relevant and may call for additional data gathering or conducting validation of the proposed operational definition for the data element, if using the proxy adds too much uncertainty to the study interpretation. FDA encourages use of measures that participants or practicing clinicians deem meaningful as potential data elements for studies using RWD. Development of endpoints or potential consideration of proxy outcomes may be warranted to address some study questions. Additionally, development of proxies for key covariates may also 846 be appropriate to address some study questions.
(3) Appropriate integration of data elements within study design and analysis 🔗
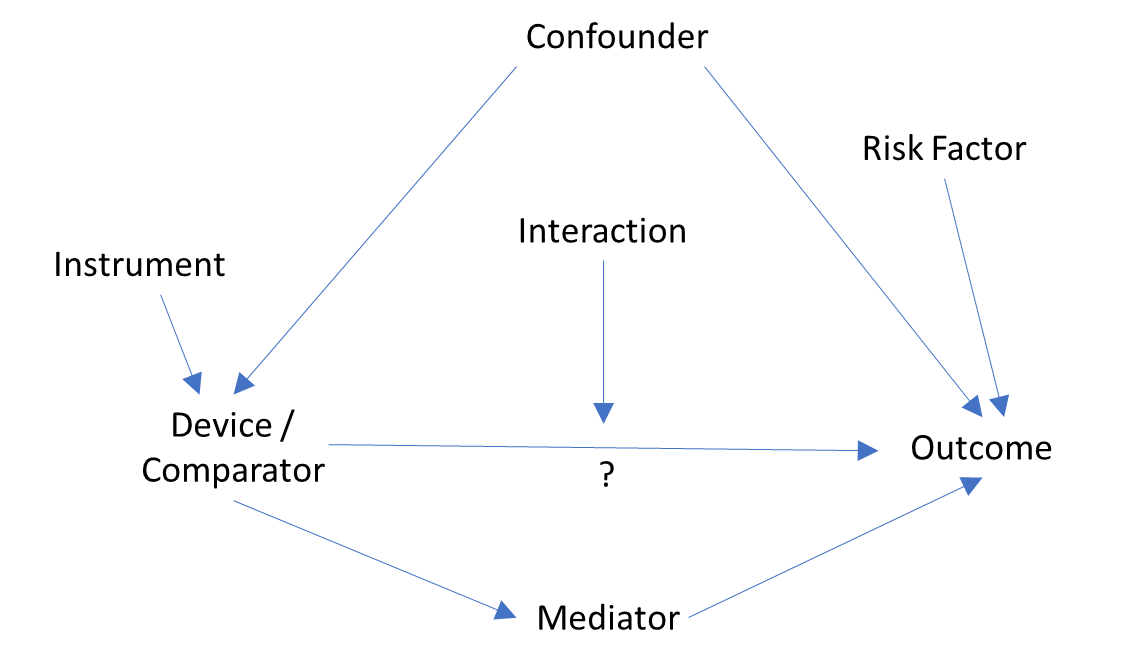
As with all clinical evidence, data elements in a study using RWD should be determined before conduct of the analysis and integrated into the study design and analysis in a manner which allows for assessment of the study question. Once the device and outcome are determined per the study question, variables which affect both the device and outcome (i.e., confounders) should be addressed within the study design and analysis to minimize bias and uncertainty. Differing levels of influence may exist and both direct and indirect influences on the device or outcome may exist for a confounder. Thus, only a subset of the confounders initially identified should be included in the study. Conversely, variables that are impacted by the device and subsequently impact the outcome (sometimes called “mediators” or “intermediate variables”) should be carefully considered before inclusion in evaluations of how the device impacts the outcome. Depending on the type of analysis, inclusion of such variables may dilute the totality of the device impact. As with all clinical studies, subgroup analyses for sex, age, racial or ethnic groups are expected because there may be differential effects of the outcome for participants across these subgroups.39 Other variables may also exhibit heterogeneity in risk of the outcome (i.e., modifiers or “interaction” terms) and stratified analyses for these variables may also be appropriate.
One way to identify which data elements fall into each of these types of variables is to generate and analyze causality diagrams. Causality diagrams (e.g., directed acyclic graphs, see Figure 3) and their subsequent assessment may provide a rationale for the design and analysis choices. Additionally, causality diagrams may provide a resource to aid discussions for a study design amongst the study team or with FDA. Covariates affecting and affected by both the exposure and outcome are noted within the causality diagram, irrespective of availability within RWD, and are assessed for potential relationships between the variables.
Figure 3. Directed acyclic graph to identify potential data elements and assess which are key for the study question
VII. Documentation for FDA Review 🔗
This section describes the documentation recommended to support the use of the RWD for generating RWE for regulatory purposes and applies to device regulatory submissions submitted to CDRH and CBER, including but not limited to, pre-submissions, 510(k)s, PMAs, BLAs, HDEs, De Novos, IDEs, post-approval study PMA supplements, 522 submissions, CLIA Waiver by Applications, and Duals.
A. Regulatory Submission Cover Letters 🔗
FDA recommends sponsors identify RWD and RWE as part of the regulatory submission cover letter to help facilitate review and internal tracking. Specifically, FDA recommends sponsors include the following elements in the cover letter for each submission that includes RWD:
- Purpose of using RWE to support the submission (see list of examples in Section IV.A.);
- Description of where the RWE fits into the totality of clinical evidence submitted (e.g., to support interpretability of the primary evidence, to establish a performance goal, to supplement clinical evidence) (see list of examples in Section IV.A.);
- Study design (i.e., type of study) using RWD to generate RWE (see list of examples in Section VI.A.);
- RWD source(s) used to generate RWE (see list of examples in Section IV.A.); and
- Specific RWD source(s) and version, including the following information, if applicable:
- Data source name;
- Data source provider;
- Version number; and
- Date of extraction and date range of data extracted.
B. Fit-For-Purpose Assessment 🔗
If sponsors include RWE in support of regulatory submissions, they should include their fit-for-purpose assessment of the relevance and reliability of the RWD to generate RWE with the following elements:
- An assessment of the key relevance and reliability factors for the study using RWD (see Section V.), which may include, but is not limited to the following:
- Data availability, linkages, timeliness (see Section V.A.);
- Data accrual, quality and integrity (see Section V.B.);
- Study purpose, specific data elements, generalizability of data, assessment of confounding, timing of data availability (see Sections V.A. and VI.B.); and
- Completeness and accuracy of study sample reflecting the target population, study design and planning (see Section V.B.2.).
In addition to the fit-for-purpose assessment of the RWD, we recommend that the sponsor provide the following contextual information regarding how the generated RWE fits into the totality of evidence:
- A description of how the RWE is being used in the totality of clinical evidence submitted to FDA;
- A summary of how the totality of the relevance and reliability of the RWD is fit-for-purpose to address the study question; and
- If unique considerations for the specific RWD source exist, sponsors should describe these considerations and how they impact the overall assessment of the data.
C. Protocol 🔗
As with traditional clinical studies, sponsors should submit the protocol as part of the regulatory submission to FDA. In studies designed to test a hypothesis, FDA recommends that sponsors finalize the protocol and analysis plan prior to reviewing the outcome data of a study and before performing the prespecified analyses. Sponsors should indicate in the regulatory submission whether or not the protocol and analysis plan were finalized prior to the analyses. In addition, individuals generating summary scores (e.g., propensity score modeling) should not have access to the outcomes within the dataset(s) used for the study. Any revisions to the protocol should be dated and time-stamped, and the rationale for each change should be provided.
Similar to protocols submitted with traditional clinical studies, sponsors should consider providing the following information in the protocol for the study generating the RWE, when the RWD or RWE is included in the regulatory submission:
- Study synopsis;
- Background and study purpose;
- Explanation of how the source data is or is not representative of the general disease/population with the condition, including sufficient previous research to interpret study results within the context of the target population, disease trajectory, and current clinical care; and
- Description of the device included in the study, including the DI portion of the UDI, if available. For devices excepted from the UDI requirements, sponsors should include the version(s) of the device.
- Study aims and objectives;
- Study design, including study period;
- Study design diagrams are suggested to clarify (1) potential study entry dates within study period and (2) assessment of all other data elements in relation to cohort entry or index date (causality diagram recommended);
- Data source, including a description of how the setting/environment(s) of data capture provided adequate continuity of care (see Section V.B.);
- Identification of any common data model structure used for housing the RWD source or for transformed study-specific RWD, if applicable.
- Data elements (conceptual and operational definitions for all), including:
- Determination of initiation, continuance, and discontinuation of device exposure, if applicable (see Section V.B.);
- Study population, including inclusion and exclusion criteria;
- Device and comparator, if included in the study;
- Outcome and endpoints; and
- Covariates.
- Statistical/data analysis plan;
- Data management and quality control plans (see Section V.B.);
- Sample size and statistical power;
- Description of human subject protections, as appropriate, including informed consent, IRB determination, deidentification plan (e.g., to remove participant identifiers from patient-generated data or device-generated data), and data confidentiality plans;
- Plans for adverse event reporting;40
- Milestones and timeline;
- Auditing and monitoring plans, as applicable;
- If validation or adjudication of data element(s) were conducted (see Section VI.B.), sponsors should include the study plan and results for validation or adjudication; and
- A copy of the data dictionary used, if one was used or developed.
D. Report 🔗
As with traditional clinical studies, sponsors should submit the report as part of the regulatory submission to FDA. The report should include the occurrence and rationale for any protocol deviations.
Additionally, in a regulatory submission with a report of a study using RWD, the study results, discussion, and conclusion should be included, including how the RWE supports the purpose of the submission. Specifically, sponsors should include the following information:
- Date of data extraction (see Section V.B.);
- All elements from the protocol, updated to reflect how the study was conducted; and
- Justification that any changes or modifications to the protocol did not affect the validity of the resulting RWE.
E. Additional Information 🔗
As with traditional clinical studies, sponsors should also provide the following information in regulatory submissions that include RWE:
- ClinicalTrials.gov National Clinical Trial (NCT) Number,41 if applicable;
- Informed consent and IRB documentation, as applicable;
- Initial and continuing IRB review and approval; and
- Initial and approved changes to informed consent.
- List of investigational sites, if any, including mailing address, contact information, and investigator name; and
- Case Report Form templates, as developed by the sponsor, if applicable (e.g., these templates may be helpful if EHR or claims data are not mapped to the dataset from the source).
Appendix A. Recommended Elements for Documentation and FDA Review 🔗
The following is an example of the recommended elements to assist sponsors and FDA in determining relevance and reliability of the RWD. The tables below summarize the recommended elements identified throughout the guidance for sponsors to document and provide in FDA submissions, and the recommended locations for where to include this information. The tables are not intended to serve as either a mandatory or exclusive checklist. Rather, the tables provide a simplified summary of the key elements that sponsors should use to assess relevance and reliability.
Table 1- Recommended RWD Relevance Elements for Submission of RWE
| Item (Linked to Section V.) | Information for Sponsors to Document (e.g., to make available for inspection) | Information for Sponsors to Provide to FDA in Submission | Recommended Location in FDA Submission |
| Determine RWD source contains sufficient detail to capture data elements and address the study question | x detailed | x (rationale) | Protocol (rationale for study question and data element definitions) |
| Assess longitudinality of data source | x | Protocol | |
| Assess continuity of care in data source | x | Protocol and report | |
| Ensure reasonable time between data collection and release for research | x | Protocol and report | |
| Consider changes in clinical practice/guidelines over time | x | x | Protocol |
| Assess timing of availability of any new (i.e., updated) data after initial data availability | x | Protocol and report | |
| Assess whether and how data from different sources can be obtained and integrated, given the potential for heterogeneity in population characteristics, clinical practices, and coding across data sources | x | Protocol | |
| If done, use of a predefined linkage methodology that is scientifically valid and accounts for differences in coding and reporting across sources | x (detailed) | x (high level) | Protocol |
| Assess adequacy of line-level linkages | x | Report | |
| Correct for redundant data, to resolve any inconsistencies, and assess the potential for missing data | x | Report | |
| Demonstrate interoperability of the linked data systems | x | Report | |
| Ensure study sample is representative and generalizable to RWD source and target population | x | Protocol and report |
Table 2- Recommended RWD Reliability Elements for Submission of RWE
| Item (Linked to Section V.) | Information for Sponsors to Document | Information for Sponsors to Provide to FDA in Submission | Recommended Location in FDA Submission |
| Establish information and descriptors about data source(s) | x | Protocol | |
| If applicable, describe defined processes, site training, support, qualified personnel for complete and accurate data collection | x | ||
| Document routine migration of data from various sources over time | x | ||
| Describe sources and technical methods used for data element capture | x | ||
| Describe methods for data retrieval and processes to minimize missing data extraction, implausible values, and data quality checks | x | ||
| If not data holder, describe level of access, attempt to gain patient-level data, and consider access for FDA | x | Protocol | |
| Describe the quality of the data captured | x (detailed) | x (high level) | Report |
| Plan and document the process of extraction, aggregation, curation, storage, and availability of data for research | x | ||
| Describe data flow from first instance to data source instance as housed by sponsor | x | ||
| Define and follow procedures for data collection and quality assurance | x | ||
| Provide assessment of completeness, accuracy, and consistency across sites and over time | x | Report | |
| Assess consistency of data capture across sites and over time; if any changes are needed (e.g., diagnostic criteria or clinical definitions), document and assess their impact on results | x (detailed) | x (high level) | Report |
| Assess missingness and out-of-range values for each data element | x | Protocol and report | |
| Ensure data elements captured are reflective of the actual patient experience (e.g., interactions with health care, disease trajectory, outcomes) | x | Protocol and report | |
| Define the auditing rules and methods used and the mitigation strategies used to reduce errors | x | ||
| Ensure study size is adequate to address the study question with statistical power and planned analyses | x | Protocol and report | |
| Document adherence to source verification procedures and data collection and recording procedures for completeness and consistency | x | ||
| Prespecify data checks and procedures to help address identified errors | x | ||
| Describe mitigations to address audit findings, including data corrections | x | ||
| Securely store data and ensure appropriate permissions/agreements for access | x | ||
| Provide documentation of previous RWD source fit-for-purpose assessment and peer-reviewed literature of RWE generation | x | Protocol | |
| Ensure adequate patient protections (e.g., privacy, ethics compliance) are in place before executing protocol | x | Protocol and report |
Appendix B. Examples Where RWE is Used 🔗
Most of the following examples are generalized from actual uses of RWD in support of FDA regulatory decision-making. These examples do not represent a comprehensive list of all potential uses or sources of RWD but do describe some situations where RWE might be used to support regulatory decision-making.For additional examples of RWE used in regulatory decisions, see the following FDA document: Examples of Real-World Evidence (RWE) Used in Medical Device Regulatory Decisions.42
Example 1: New or Expanded Indications for Use
An implanted device, which was available outside the US (OUS), used RWE as the primary clinical evidence to support the original PMA submitted to FDA. RWD from an OUS registry in one country, which included nearly 300 patients with more than two years of follow-up, was compared against a performance goal (PG) for device effectiveness. The PG was derived from a prospectively defined, systematic meta-analysis of available published literature and registry data for a control device legally marketed in the United States. The study prospectively evaluated the functional outcome and patient satisfaction for multiple devices. The safety assessment was based on a comparison of the serious device related adverse event rates for the subject device to the rates extracted from the registry and the same literature studies used to derive the PG. These analyses served as the primary basis supporting approval of the PMA. In this example, an IDE was not needed for the study as the clinical data for the subject device was obtained from extant data in an OUS registry.
Should a manufacturer wish to expand indications, this type of RWD might be used to generate sufficient evidentiary support, depending on the specific devices, indications, and analyses.
Another example is an implanted device that was available on the US market for several years for one indication for use. The sponsor submitted a PMA panel-track supplement to support expanding indications for this device. Supplemental clinical evidence for safety of the indication expansion was provided from the sponsor’s patient database, with more than 36,000 patients, linked with Medicare claims. The RWE characterized the 12-month safety profile of patients with the specific condition (i.e., new indication) implanted with the device and assessed the safety among patients receiving the implant and diagnosed with the specific condition compared to patients implanted with the device that did not have the respective condition. The sponsor’s patient database and Medicare claims were linked using probabilistic methods (i.e., based on date of birth, implant date, implant clinic). A systematic review of literature was also conducted to identify diagnostic codes associated with safety outcomes. In this example, an IDE was not needed for the study as the clinical data for the subject device was obtained from extant data.
Example 2: 522 Submissions
The manufacturer of a class II designated software-only device was subject to a postmarket surveillance order for this device under section 52243 of the FD&C Act, as it met the statutory criteria that its failure would be reasonably likely to have serious adverse health consequences, and it also was expected to have significant use in pediatric populations.
Following discussions with the FDA, the postmarket surveillance study for this software-only device was a single-arm, prospective study designed to assess the 12-month safety in a real-world setting and to support the continued assessment of the software for its intended use. It included both pediatric and adult populations. The outcomes were evaluated versus a comparator group, which was determined using evidence from a systematic literature review of similar use in the intended use population. Device-related adverse events were recorded electronically from both inbound reports from customers and assessed through outreach to patients. The safety endpoints included the risk for adverse events as extracted in the literature review. Besides the safety and effectiveness endpoints, the study also included a secondary endpoint of patient-reported satisfaction with and trust in the software-only device. At the end of the study, the device had met its endpoints, and there were no unanticipated adverse device effects.
Example 3: Control group
A sponsor submitted a PMA panel track supplement to support an indication expansion supported by a single-arm study compared to a control group of patients enrolled in a US registry receiving alternative devices for the new disease condition. The alternative devices were identified through the use of UDIs. To account for potential differences between the device and control groups, the sponsor performed a propensity-score adjusted analysis using 20 pre-specified variables. The propensity score results were reviewed by FDA before the sponsor performed the outcome analysis. Sensitivity analyses were conducted to assess the impact of missing data. The registry did not routinely collect all relevant data for the control group, therefore, additional information for patients in the registry was collected from extant EHRs. The results of these analyses demonstrated that the success criteria were achieved.
Along with analyses of serious adverse events from the sponsor’s global study, this RWE was the primary basis for supporting approval of the PMA supplement.
Example 4: Supplementary Data
A sponsor submitted a De Novo request for a device. Although the sponsor conducted a prospective, repeated measures, single arm study as the primary clinical evidence to support safety and effectiveness for their De Novo request, RWD from a chart review at a single site was used as supplemental evidence. The overall objective of the study using RWD was to confirm the performance of the subject device as part of the standard of care and to further investigate trends in treatment outcomes for patients with different severities of the disease. The RWE generated from this data provided clinical practice results for a patient population that was not included in one of the clinical studies and demonstrated consistent outcomes with the prospective single arm study.
Example 5: RWE Obtained from Use of EUA device
In response to the COVID-19 outbreak, FDA authorized the emergency use of certain devices under section 564 of the FD&C Act.44 A sponsor of a serology test authorized for emergency use used RWD from a patient registry to support the expansion of the intended use (i.e., asymptomatic testing when authorized only for symptomatic testing) in a subsequent Dual 510(k) and CLIA Waiver by Application. The RWD from the patient registry supplemented data from a more traditional clinical study and from peer-reviewed literature. Following Agency review, FDA considered the RWE to be valid scientific evidence; the RWD was fit-for-purpose and potential sources of bias were minimized. The RWD was fit-for-purpose as it included appropriate data elements that were captured in the data and was appropriately linked to the specific assay in question (e.g., coding scheme using LOINC or Systematized Nomenclature of Medicine Clinical Terms [SNOMED CT]) in addition to information about the setting in which the assay was performed and the operators who performed the test.45 Additionally, it was important that the sponsor ensured that the DI, patient identifier (ensuring appropriate patient population and rationale for any exclusions (e.g., vaccination status)), relevant demographic and clinical information, and their interrelationships were available in a single database or commonly available RWD sources. In this example, the sponsor accounted for the use of different instruments in generating the results as well as the use of different controls that make it difficult to show equivalent performance or understand the comparator used.
Footnotes 🔗
-
For more information on regulatory submissions for medical devices, see Sections III. and VII. ↩
-
For the purposes of this draft guidance, an “EHR" is an electronic record of health-related information on an individual that conforms to nationally recognized/utilized interoperability standards and that can be created, managed, and consulted by authorized clinicians and staff across more than one health care organization. Definition adapted from The National Alliance for Health Information Technology Report to the Office of the National Coordinator for Health Information Technology on Defining Key Health Information Technology Terms April 28, 2008, available at https://webarchive.library.unt.edu/eot2008/20080920183033/http:/www.hhs.gov/healthit/documents/m20080603/10_2_hit_terms.pdf ↩
-
3 See FDA’s guidance, “Requests for Feedback and Meetings for Medical Device Submissions: The Q-Submission Program,” available at https://www.fda.gov/regulatory-information/search-fda-guidance-documents/requests-feedback-and-meetings-medical-device-submissions-q-submission-program ↩
-
For the purposes of this draft guidance, we use the term “clinical studies” in this guidance as a broad term to capture clinical research regarding the safety or effectiveness of a device, regardless of study design. We use the term “traditional clinical studies” to refer to clinical studies that do not utilize RWD. ↩
-
Under 21 CFR 860.7(c)(2), “valid scientific evidence” is considered “evidence from well-controlled investigations, partially controlled studies, studies and objective trials without matched controls, well-documented case histories conducted by qualified experts, and reports of significant human experience with a marketed device, from which it can fairly and responsibly be concluded by qualified experts that there is reasonable assurance of the safety and effectiveness of a device under its conditions of use. The evidence required may vary according to the characteristics of the device, its conditions of use, the existence and adequacy of warnings and other restrictions, and the extent of experience with its use.” ↩
-
For the purposes of this draft guidance, “patient experience data” means data that are (1) collected by any persons (including patients, family members and caregivers of patients, patient advocacy organizations, disease research foundations, researchers, and drug manufacturers); and (2) are intended to provide information about patients’ experiences with a disease or condition, including (A) the impact of such disease or condition, or a related therapy, on patients’ lives; and (B) patient preferences with respect to treatment of such disease or condition. See section 3001 of the 21st Century Cures Act, Pub. L. No. 114-255 (December 2016). ↩
-
For the purposes of this draft guidance, “medical device registry” means an organized system with a primary aim to increase the knowledge on medical devices contributing to improve the quality of patient care that continuously collects relevant data, evaluates meaningful outcomes and comprehensively covers the population defined by exposure to particular device(s) at a reasonably generalizable scale (e.g., international, national, regional, and health system). Definition is cited from the IMDRF document “Principles of International System of Registries Linked to Other Data Sources and Tools,” available at https://www.imdrf.org/documents/principles-international-system-registries-linked-other-data-sources-and-tools ↩
-
For more information, see FDA’s guidance “The Least Burdensome Provisions: Concept and Principles,” available at https://www.fda.gov/regulatory-information/search-fda-guidance-documents/least-burdensome-provisions-concept-and-principles ↩
-
For more information, see FDA’s guidance “Balancing Premarket and Postmarket Data Collection for Devices Subject to Premarket Approval,” available at https://www.fda.gov/regulatory-information/search-fda-guidance-documents/balancing-premarket-and-postmarket-data-collection-devices-subject-premarket-approval ↩
-
Available at https://www.fda.gov/regulatory-information/search-fda-guidance-documents/use-real-world-evidence-support-regulatory-decision-making-medical-devices ↩
-
Available at https://www.fda.gov/media/158308/download ↩
-
This guidance does not apply to drugs and biological products. For information on the RWE program for drugs and biological products, see https://www.fda.gov/science-research/real-world-evidence/center-biologics-evaluation-and-research-center-drug-evaluation-and-research-real-world-evidence ↩
-
For the purposes of this draft guidance, “administrative claims data” means claims data that arise from a person’s use of the health care system and reimbursement of health care providers for that care. Definition adapted from Strom et al., Textbook of Pharmacoepidemiology 6th ed. 2022, page 137. ↩
-
Generally, FDA does not consider published literature to be RWD. Literature may report data from an RWD source, in which case sponsors should specify the RWD source type (e.g., if a journal article presents a retrospective analysis of EHRs, the RWD source should be specified as EHR in the cover sheet). ↩
-
For the purposes of this draft guidance, “registry” means an organized system that uses observational study methods to collect uniform data (clinical and other) to evaluate specified outcomes for a population defined by a particular disease, condition, or exposure, and that serves one or more stated scientific, clinical, or policy purposes. Definition adapted from the Agency for Healthcare Research and Quality’s (AHRQ’s) “Registries for Evaluating Patient Outcomes: A User’s Guide,” available at https://effectivehealthcare.ahrq.gov/products/registries-guide-4th-edition/users-guide ↩
-
The use of the term “patient-generated data” is consistent with the use of this term in the “Framework for FDA’s Real-World Evidence Program” document, available at https://www.fda.gov/media/120060/download. Patientgenerated data includes patient-generated health data. For the purposes of this draft guidance, “patient-generated health data” means health-related data created, recorded, or gathered by or from patients, family members, or other caregivers to help address a health concern. Definition adapted from https://www.healthit.gov/topic/scientific-initiatives/pcor/patient-generated-health-data-pghd ↩
-
For the purposes of this draft guidance, “digital health technology” means a system that uses computing platforms, connectivity, software, and/or sensors for health care and related uses. These technologies span a wide range of uses, from applications in general wellness to applications as a medical device. They include technologies intended for use as a medical product, in a medical product, or as an adjunct to other medical products (devices, drugs, and biologics). They may also be used to develop or study medical products. ↩
-
In 2019, an outbreak of respiratory disease caused by a novel coronavirus began. The virus has been named “SARS-CoV-2,” and the disease it causes has been named “Coronavirus Disease 2019” (COVID-19). On January 31, 2020, the Secretary of Health and Human Services (HHS) issued a declaration of a Public Health Emergency (PHE) related to COVID-19 in accordance with section 319 of the Public Health Service Act (PHS Act) (hereinafter referred to as “section 319 PHE declaration”) and mobilized the Operating Divisions of HHS. In addition, on March 13, 2020, the President declared a national emergency in response to COVID-19. The section 319 PHE declaration related to COVID-19 expired on May 11, 2023. ↩
-
For more information on Bayesian trails, see FDA’s guidance, “Guidance for the Use of Bayesian Statistics in Medical Device Clinical Trials,” available at https://www.fda.gov/regulatory-information/search-fda-guidance-documents/guidance-use-bayesian-statistics-medical-device-clinical-trials-pdf-version ↩
-
For more information on CLIA waiver by applications, see FDA’s guidance, “Recommendations for Clinical Laboratory Improvement Amendments of 1988 (CLIA) Waiver Applications for Manufacturers of In Vitro Diagnostic Devices,” available at https://www.fda.gov/regulatory-information/search-fda-guidance-documents/recommendations-clinical-laboratory-improvement-amendments-1988-clia-waiver-applications ↩
-
For more information on the Dual 510(k) and CLIA Waiver by Application pathway, see FDA’s guidance, “Recommendations for Dual 510(k) and CLIA Waiver by Application Studies,” available at https://www.fda.gov/regulatory-information/search-fda-guidance-documents/recommendations-dual-510k-and-clia-waiver-application-studies. Dual De Novo classification requests and CLIA Waiver by Application for certain devices (see section 3301 of FDORA) should also consult this guidance. ↩
-
See FDA’s guidance, “General/Specific Intended Use,” available at https://www.fda.gov/regulatory-information/search-fda-guidance-documents/generalspecific-intended-use-guidance-industry ↩
-
See FDA’s guidance, “Balancing Premarket and Postmarket Data Collection for Devices Subject to Premarket Approval,” available at https://www.fda.gov/regulatory-information/search-fda-guidance-documents/balancing-premarket-and-postmarket-data-collection-devices-subject-premarket-approval ↩
-
See 21 CFR 812.2(a). ↩
-
See 21 CFR 812.3(h). ↩
-
Under sections 564(a)(2)(A) and 564(a)(4)(D) of the FD&C Act, an unapproved product is one that “is not approved, licensed, or cleared for commercial distribution under section 505, 510(k), 512, or 515 of [the FD&C] Act or section 351 of the [PHS] Act or conditionally approved under section 571 of [the FD&C] Act.” ↩
-
See, for example, 21 CFR 812.28(a)(1), which defines good clinical practice in the context of device investigations conducted outside the United States. ↩
-
For example, see the following FDA guidances: Guidance for the Use of Bayesian Statistics in Medical Device Clinical Trials; “Evaluation of Sex-Specific Data in Medical Device Clinical Studies,” available at https://www.fda.gov/regulatory-information/search-fda-guidance-documents/evaluation-sex-specific-data-medical-device-clinical-studies-guidance-industry-and-food-and-drug ; and “Evaluation and Reporting of Age-, Race-, and Ethnicity-Specific Data in Medical Device Clinical Studies,” available at https://www.fda.gov/regulatory-information/search-fda-guidance-documents/evaluation-and-reporting-age-race-and-ethnicity-specific-data-medical-device-clinical-studies ↩
-
The exposure is the variable or data element whose causal effect is estimated in a study. For many studies supporting a regulatory submission, the exposure is the medical device. For some studies, especially those providing supportive clinical evidence, another exposure may be assessed to provide context for the use, safety, or effectiveness of the medical device within clinical practice. ↩
-
The device identifier is a mandatory, fixed portion of a UDI that identifies the specific version or model of a device and the labeler of that device. 21 CFR 830.3. For more information regarding UDI, see FDA’s webpage https://www.fda.gov/medical-devices/unique-device-identification-system-udi-system/udi-rule-guidances-training-and-other-resources ↩
-
For class I devices, the universal product code (UPC) may serve as the UDI (21 CFR 801.40(d)). In these instances, the UPC should be included. ↩
-
For the purposes of this guidance, “continuum of care” refers to the extent of the individual’s pertinent health data which is captured across settings/environments of care is represented in the RWD source ↩
-
See An Overview of Record Linkage Methods - Linking Data for Health Services Research - NCBI Bookshelf, available at https://www.ncbi.nlm.nih.gov/books/NBK253312/ ↩
-
More information on PCORI Conduct of Registry Studies is available at https://www.pcori.org/sites/default/files/Standards-in-the-Conduct-of-Registry-Studies-for-Patient-Centered-Outcomes-Research1.pdf ↩
-
For more information on documentation for FDA review, see Section VII. and Appendix A. ↩
-
For the purposes of this draft guidance, the “first instance” is considered to be the data as initially available to the sponsor. For example, if the raw data from an EHR is available, then the first instance is at the point of capture for the data element. However, if the sponsor only has access to curated data or a specific dataset, then the “first instance” is the initially ingested data by the sponsor, even though it is not the original data collected. ↩
-
Available at https://www.fda.gov/regulatory-information/search-fda-guidance-documents/design-considerations-pivotal-clinical-investigations-medical-devices ↩
-
Such uses of RWD, while beyond the scope of this draft guidance, may be valuable for describing the clinical context in which the device will be used, to support a diversity action plan. See section 520(g)(9) of the FD&C Act for more information on diversity action plans for medical devices. ↩
-
See FDA’s guidances, including, Evaluation of Sex-Specific Data in Medical Device Clinical Studies; “Collection of Race and Ethnicity Data in Clinical Trials,” available at https://www.fda.gov/regulatory-information/search-fda-guidance-documents/collection-race-and-ethnicity-data-clinical-trials ; and Evaluation and Reporting of Age-, Race-, and Ethnicity-Specific Data in Medical Device Clinical Studies ↩
-
See 21 CFR part 803. ↩
-
ClinicalTrials.gov assigns a unique NCT number to each clinical study registered on their webpage. See https://clinicaltrials.gov/ for more information. ↩
-
Available at: https://www.fda.gov/media/146258/download ↩
-
FDA has issued a series of postmarket surveillance orders, related to investigating patient safety issues for a type of class II device, under the authority of section 522 of the FD&C Act. These 522 orders cover multiple devices from different manufacturers that are similar in intended use, design, and other characteristics, such that the surveillance questions are identical. ↩
-
For more information on FDA’s emergency use authorities under section 564 of the FD&C Act, see FDA’s guidance “Emergency Use Authorization of Medical Products and Related Authorities,” available at https://www.fda.gov/regulatory-information/search-fda-guidance-documents/emergency-use-authorization-medical-products-and-related-authorities ↩
-
For more information on the Dual 510(k) and CLIA Waiver by Application pathway, see FDA’s guidance, Recommendations for Dual 510(k) and CLIA Waiver by Application Studies. ↩
