
Innolitics introduction 🔗
About this Transcript 🔗
This document is a transcript of an official FDA (or IMDRF) guidance document. We transcribe the official PDFs into HTML so that we can share links to particular sections of the guidance when communicating internally and with our clients. We do our best to be accurate and have a thorough review process, but occasionally mistakes slip through. If you notice a typo, please email a screenshot of it to Mihajlo at mgrcic@innolitics.com so we can fix it.
Preamble 🔗
Document issued on December 16, 2019.
Draft document issued on May 1, 2003.
For questions about this document regarding CDRH-regulated devices, contact the Office of Regulatory Programs at 301-796-5640. For questions about this document regarding CBER-regulated devices, contact the Office of Communication, Outreach, and Development (OCOD) at 1-800-835-4709 or 240-402-8010, or by email at ocod@fda.hhs.gov.
Contains non-binding guidance.
I. INTRODUCTION 🔗
As discussed in more detail below, the PMA regulation (21 CFR 814.42(e)) identifies the criteria that, if not met, may serve as a basis for refusing to file a PMA. This guidance is intended to be used by FDA staff and the device industry to help elucidate the broad preclinical and clinical issues that need to be addressed in a PMA and the key decisions to be made during the filing process.
Focusing the Agency’s review resources on complete applications will provide a more efficient approach to ensuring that safe and effective medical devices reach patients as quickly as possible. Moreover, with the enactment of the Medical Device User Fee and Modernization Act of 2002 (MDUFMA), the Medical Device User Fee Amendments of 2007 (MDUFA II), the Medical Device User Fee Amendments of 2012 (MDUFA III), and the Medical Device User Fee Amendments of 2017,1 FDA agreed to performance goals based on the timeliness of reviews. Acceptance review therefore takes on additional importance in both encouraging quality applications from PMA applicants and allowing the Agency to appropriately concentrate resources on complete applications.
Therefore, we have modified the PMA filing guidance and checklist. We have separated the criteria for PMA filing into 1) acceptance criteria and 2) filing criteria. Acceptance review involves assessment of the completeness of the application, and informing the applicant in a written response within the first 15 calendar days of receipt2 of the application by the document control center (DCC) whether any elements are missing, and if so, identifying the missing element(s). In order to enhance the consistency of our acceptance and filing decisions and to help applicants better understand the types of information FDA needs to conduct a substantive review of a PMA, this guidance and associated checklist clarify the necessary elements and contents of a complete PMA application. The process we outline is applicable to all devices reviewed in a PMA application and has been compiled into a checklist for use by FDA review staff.
FDA staff and industry should note that this guidance is not significantly different from the previous PMA filing checklist and guidance document, as the PMA filing criteria defined in the regulation have not changed. The “preliminary questions” remain the same and the “filing review questions” have been separated into “acceptance decision questions” (i.e., whether the file is administratively complete) and “filing decision questions” (i.e., determine the basic adequacy of the technical elements of the PMA). In the 2003 PMA Filing Guidance, we stated that delayed submission of the manufacturing section would not preclude filing a PMA, and, if this section was not included in the original PMA application, recommended submitting this section within 90 days. However, delayed submission of the manufacturing section has rarely occurred in recent years, and in many cases this section is submitted prior to other sections of the PMA, as part of a modular PMA application. Therefore, we are now including the manufacturing section in the checklist for a complete PMA application for an original PMA or a panel-track supplement (with a new manufacturing site or substantially different manufacturing procedures).
FDA requires all applicants to provide one electronic copy (eCopy) of the PMA application. The eCopy must be accompanied by a single paper copy of your signed cover letter. FDA has issued guidance3 to implement Section 745A(b) of the FD&C Act, added by section 1136 of FDASIA, which provides statutory authority to require an eCopy for most submissions, including original PMAs and PMA supplements. With the implementation of this provision, a valid eCopy is required in order for a PMA or PMA supplement to be processed and for the acceptance review to begin.
FDA's guidance documents, including this guidance, do not establish legally enforceable responsibilities. Instead, guidances describe the Agency's current thinking on a topic and should be viewed only as recommendations, unless specific regulatory or statutory requirements are cited. The use of the word should in Agency guidance documents means that something is suggested or recommended, but not required.
II. PURPOSE 🔗
The purpose of the PMA acceptance and filing reviews is to make a threshold determination about whether an application is administratively complete for the Agency to undertake a substantive review. The PMA regulation (21 CFR 814.42(e)) states that FDA may refuse to file a PMA if any of the following applies:
(1) The PMA is incomplete because it does not on its face contain all the information required under section 515(c)(1)(A)-(G) of the FD&C Act.
(2) The PMA does not contain each of the items required under section 814.20 and justification for omission of any item is inadequate.
(3) The applicant has a pending premarket notification under section 510(k) of the FD&C Act with respect to the same device, and FDA has not determined whether the device falls within the scope of section 814.1(c).
(4) The PMA contains a false statement of material fact.
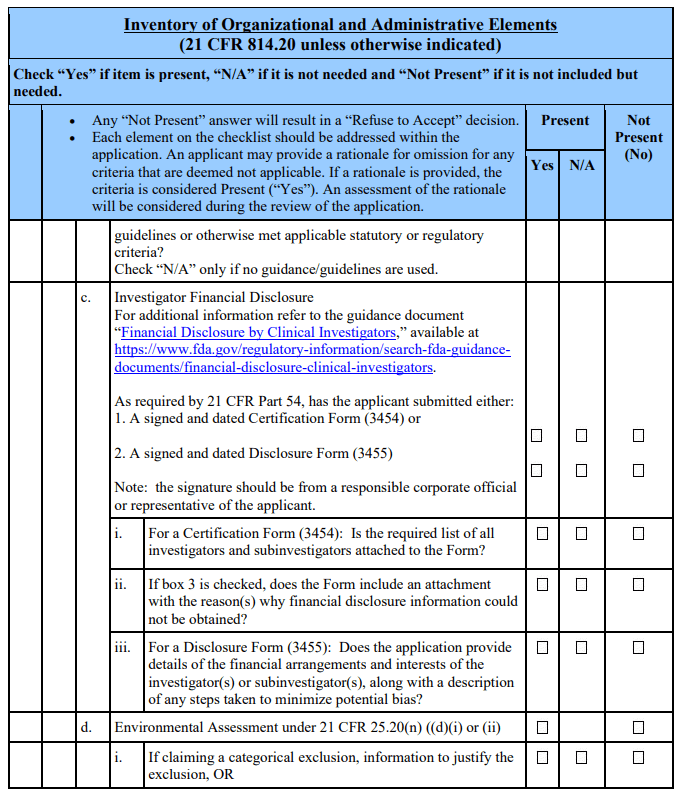
(5) The PMA is not accompanied by a statement of either certification or disclosure as required by 21 CFR Part 54.
Section 814.20 of the regulation further specifies that PMAs must include, among other things, “technical sections which shall contain data and information in sufficient detail to permit FDA to determine whether to approve or deny approval of the application” (21 CFR 814.20(b)(6)). FDA staff has frequently expressed the need for more specific guidance in applying this regulatory standard to the PMA application filing decision-making process.
The goal of this document is to clarify the criteria for accepting and filing a PMA, thereby enhancing the consistency of our acceptance and filing decisions. The decision-making process presented in this document is captured in “Checklists for Acceptance and Filing of PMAs,” (see Appendix A). FDA staff will use these checklists during the acceptance and filing review processes.
III. SCOPE 🔗
The information presented in this document is intended to provide FDA staff with a clear, consistent approach to making acceptance and filing decisions on original PMA applications4 and panel-track PMA supplements. FDA’s decision to accept and/or file a PMA does not imply that the data provided in the PMA demonstrate reasonable assurance of the safety and effectiveness of your device or assure approval of the PMA.
In addition, it should be noted that this document is focused on the regulatory and scientific criteria for making an “Accept” or “Refuse to Accept” (RTA) decision as well as “File” or “Not File” decision for a PMA. It specifically does not alter the following administrative aspects of the PMA filing process: the time frame for the filing review phase (i.e., 45 days which includes the time spent conducting the acceptance review); the processes for document tracking, distribution, and handling; and the procedures for assembling the review team and setting up the filing meeting.
This document does not discuss the statutory criteria for Breakthrough Device designation.5 Information pertaining to this designation can be found in the guidance “Breakthrough Devices Program”6 published on December 18, 2018.
This document does not address the monetary aspects or the MDUFA goals associated with PMAs. For information pertaining to the fees and payment procedures for submission of a PMA, please refer to the Medical Device User Fees webpage.7
IV. Q-Submission Interaction 🔗
Prior to interacting with review staff, applicants should consult CDRH’s Division of Industry and Consumer Education (DICE) or CBER’s Manufacturers Assistance and Technical Training Branch for general information regarding the PMA regulations. Before submitting a PMA, we encourage applicants to interact with FDA review staff. Such Q-submission interaction is an important way of improving the quality and completeness of a PMA. A face to face meeting with FDA staff may be useful for applicants prior to preparing the PMA to discuss issues related to their specific device and PMA. For additional information regarding the Q-Submission Program, please refer to the guidance “Requests for Feedback on Medical Device Submissions: The Q-Submission Program.”8
In addition, CDRH’s Device Advice,9 as well as other applicable CDRH device-specific and cross-cutting guidance documents,10 provide valuable information for preparing PMAs.
V. Basic Review Policies and Procedures 🔗
Review policies for acceptance
To facilitate a more efficient review process, FDA staff will conduct an acceptance review of all original PMAs and panel-track PMA supplements11 based on objective criteria using the Checklist for Acceptance Review (see Appendix A) to ensure that the PMA is administratively complete. In order for the application to be accepted, all organizational and administrative elements should be present or a rationale should be provided for those elements determined by the applicant to be not applicable. The acceptance review should be conducted and completed within 15 calendar days of the Agency receiving the PMA application. An acceptance review will only begin for PMAs for which the appropriate user fee has been paid and a validated eCopy has been received.12
If the application contains all of the information outlined in the checklist, FDA staff should notify the applicant electronically that it has been “Accepted” and proceed to the filing review. Should FDA fail to complete the acceptance review within 15 calendar days, the application should be considered accepted, the applicant should be notified electronically, and FDA should commence with the filing review.In the case of a government closure during the 15-day review period, the review period may be extended by a comparable number of business days that the FDA buildings are closed. If the applicant receives an automated notice that the acceptance review was not completed because the acceptance review period has exceeded 15 days, FDA may send a correction notice to the applicant. In such cases, if the application is accepted and filed, the extended acceptance review period would not otherwise impact the performance goal for a MDUFA decision on the application.
If one or more of the items on the Inventory of Organizational and Administrative Elements within the Acceptance Checklist are not present, the lead reviewer conducting the acceptance review should obtain division concurrence that the application should be designated “RTA” and notify the designated PMA contact person electronically that the application has not been accepted. FDA staff should also provide the applicant with a copy of the completed acceptance checklist indicating which item(s) are the bases for the RTA designation.
The PMA applicant may respond to the RTA notification by providing the missing information identified in the checklist. The applicant should submit this information to be included in the file (i.e., as an amendment) under the originally assigned PMA number. A new application and new user fee are not necessary. Nor should the applicant re-send the entire PMA application, unless FDA determines otherwise (e.g., because the majority of the application was not in English, or the application pages were not numbered). It is sufficient to submit and address only the information requested per the acceptance checklist.
Upon receipt of the newly submitted information, FDA staff should conduct the acceptance screening again following the same procedure within 15 calendar days of receipt. If the application is still found to be incomplete, FDA staff should notify the contact person and provide the new checklist indicating the missing item(s). This cycle will continue until the PMA is found to be administratively complete.
Review policies for filing
Once the application is found to be administratively complete, FDA staff should notify the applicant that the PMA has been accepted and begin the filing review according to the Checklist for Filing Review. The objective of the filing review is to determine the basic adequacy of the technical elements of the PMA. In order for the application to be filed, the application should be sufficiently complete to permit a substantive review. Once the filing review is complete, staff will notify the applicant electronically within 45 calendar days of receipt whether the PMA has been “Filed” or “Not Filed.” See 21 CFR 814.42(a). If the PMA has been “Filed,” the agency will identify the date of receipt of the PMA or of the amendment to the PMA that enabled FDA to file the PMA.
The PMA applicant may respond to the “Not Filed” notification by providing the missing information identified in the letter. The applicant should submit this information to be included in the file (i.e., as an amendment) under the originally assigned PMA number. Upon receipt of the newly submitted information, FDA staff should conduct the filing review again following the same procedure within 45 calendar days of receipt.
During the filing review, review staff may ask for any information that should have resulted in an RTA designation during the acceptance review. Likewise, once the application has been filed, FDA may ask for any information during the substantive review that may have been unintentionally overlooked during the acceptance or filing reviews.
FDA Review Clock
As explained in section VIII.D. of the commitment letter for MDUFA IV referenced in Title II of FDASIA, Public Law 112-114, “FDA days begin on the date of receipt of the submission or of the amendment to the submission that enables the submission to be accepted (510(k)) or filed (PMA).”13 Since the PMA acceptance criteria are a subset of the PMA filing criteria under 21 CFR 814.42, an application that is “Not Accepted” is not one that enables the application to be filed. Thus, the FDA review clock does not start when an application is placed on eCopy or user fee hold or is designated “Not Accepted” or “Not Filed.” Once FDA has both “Accepted” and “Filed” an application, the FDA review clock begins as of the date of receipt of the most recent application or amendment that made the PMA complete and on which the FDA based its “Accepted” and “Filed” decisions. This date will not change even if FDA later requests information it should have requested during acceptance or filing review.
VI. Combination Product Administrative Items 🔗
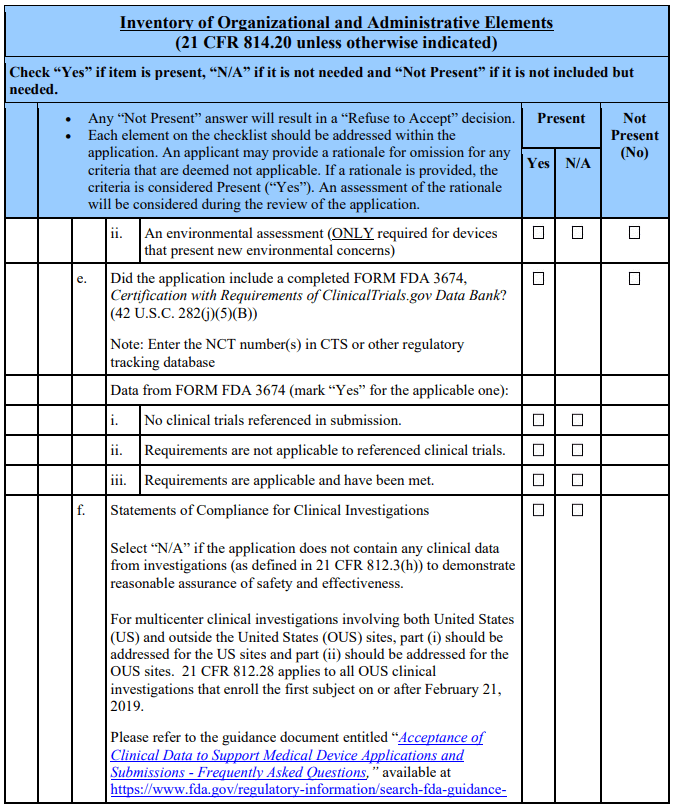
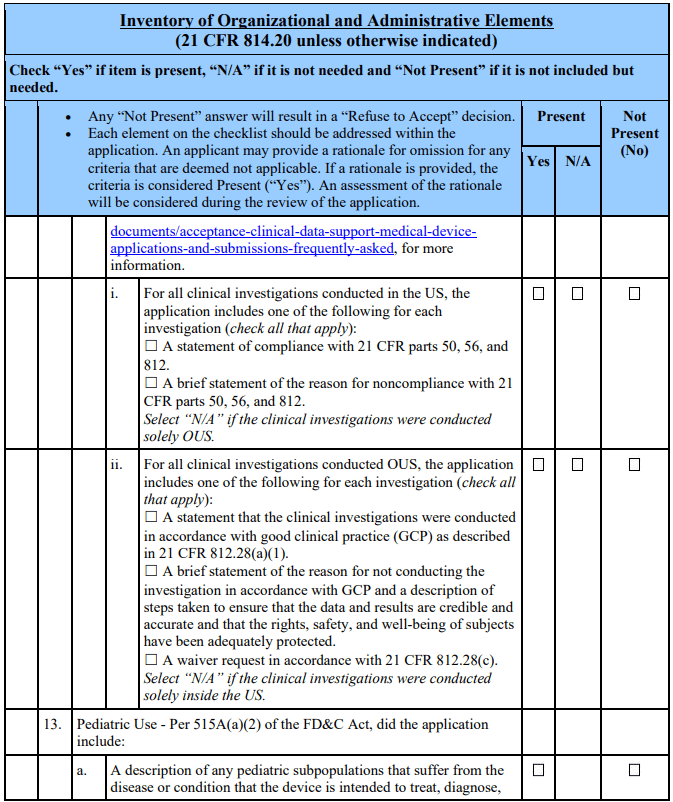
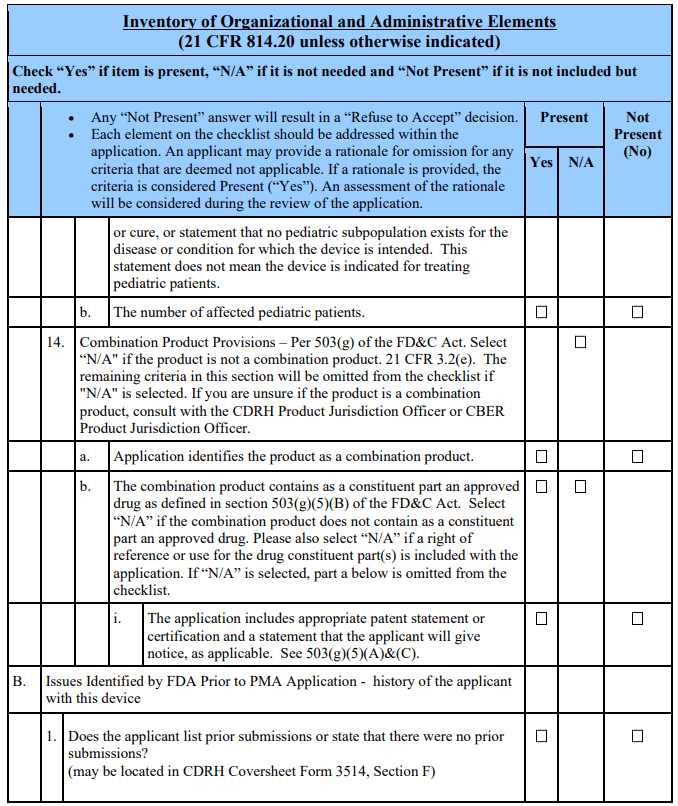
The 21st Century Cures Act, which amended section 503(g) of the FD&C Act, requires applicants seeking action on a combination product to identify the product as such [§503(g)(8)(C)(v)]. Additionally, per the amended section 503(g)(5), applications for device-led, device-drug combination products must include the patent certification or statement as described in section 505(b)(2) and provide notice as described in section 505(b)(3) if the combination product contains as a constituent part an approved drug. See section 503(g)(5)(A). Applicants of products that are not combination products, as defined in 21 CFR 3.2(e), should mark N/A to question 14 of the acceptance checklist and omit this section pertaining to combination products.
Applicants of Combination Products That Do Not Contain as a Constituent Part an Approved Drug
If the combination products do not include as a constituent part an approved drug as defined in section 503(g)(5)(B), applicants of device-led, device-drug combination products should mark “N/A” for question 14b.
Applicants of Combination Products That Contain as a Constituent Part an Approved Drug Applicants of combination products containing as a constituent part an approved drug should address question 14 by including patent information. For each relevant patent, the applicant should include certification to one of the following certifications:
i. That such patent information has not been filed (505(b)(2)(A)(i)).
ii. That such patent has expired (505(b)(2)(A)(ii)).
iii. The date on which the patent will expire (505(b)(2)(A)(iii)).
iv. That such patent is invalid or will not be infringed by the manufacture, use, or sale of the drug constituent part for which this application is made (505(b)(2)(A)(iv)).
However, for a method of use patent which does not claim a use for which the applicant is seeking approval, the applicant should include a statement per section 505(b)(2)(B) that the method of use patent does not claim such a use.
Applicants including a certification under paragraph iv (505(b)(2)(A)(iv)) should also certify that they will provide notice to the owner of the patent(s) and the holder of the approved application that lists the patent(s) that is/are being challenged. The process for giving notice is provided in section 505(b)(3) of the FD&C Act. Applicants should submit to FDA documentation of the date of receipt of notice by holder of the approved application and patent(s) owner.
VII. Acceptance and Filing Review Principles 🔗
In order to use this guidance appropriately, FDA staff should review the following basic principles in bold followed by a description of FDA’s review policies and procedures. These principles, and the objective criteria outlined in the Acceptance and Filing Checklists, inform FDA’s PMA acceptance and filing decisions.
The contents of the PMA should allow the substantive review to proceed
The PMA must contain the basic administrative and scientific elements listed in 21 CFR 814.20.
The specific questions in the acceptance and filing checklists are intended to help FDA ensure that the PMA contents are not so disorganized or incomplete so as to prevent the review team from proceeding with a substantive review of the application.
The acceptance decision and filing decision should not be based on a substantive review of the data and information in the PMA
The acceptance review and filing review are conducted to ensure that the PMA is administratively complete and to determine the basic adequacy of the technical elements of the PMA, respectively. Notably, in determining whether a PMA should be accepted and filed, the submitted information should not be evaluated to determine whether there is a reasonable assurance of safety and effectiveness. The checklist is a tool to ensure that the application contains the necessary information in order to conduct a substantive review (i.e., FDA should not designate an application “Refuse to Accept” or refuse to file a PMA because we have reviewed the data and believe that the application is ultimately not approvable). Subsequently, the substantive review of the PMA will evaluate the quality of the content and lead to a decision regarding the safety and effectiveness of the PMA product.
Concerns identified by the Agency during the acceptance or filing review regarding results and outcomes of nonclinical and clinical studies would not preclude acceptance or filing. Examples of information that would typically fall into this category include:
- demographic information for the study population
- conclusions regarding statistical analyses
- report or assessment of protocol deviations
- reports of device failures or malfunctions.
Staff should determine whether the applicant provided a justification for any alternative approach
If the applicant believes any criteria in the checklist are not applicable, it should explain its rationale. Likewise, the applicant should provide a rationale for any deviation from a device-specific or cross-cutting guidance document or FDA-recognized standard. It is FDA’s expectation that any item in the checklist that is missing will be addressed with a rationale explaining why it is not applicable and that any deviations will be explained. If a justification to omit certain information or for taking an alternative approach is provided, FDA will consider the adequacy of that justification or alternative approach during substantive review of the application.A given criterion in the checklist will be considered “Not Present” if the application fails to include either the information requested or a rationale for omission. See Acceptance Review section below for further explanation.
PMA acceptance and filing reviews
The decision to “Accept” an application or designate it “Refuse to Accept” should be made by the lead reviewer with concurrence from the immediate supervisor or designee. The decision to “File” or “Not File” a PMA should be made at the division level in collaboration with the PMA review team (e.g., the medical officer and statistician) and the appropriate managers in the reviewing division(s).
VIII. The Checklist – Preliminary Questions 🔗
Within 15 calendar days of receipt of the PMA by DCC and prior to the filing review, the PMA lead reviewer should answer the preliminary questions below, and complete the Inventory of Organizational and Administrative Elements within the Administrative Checklist to make an Acceptance Decision.
The preliminary questions are included on the first page of the “Checklists for Accepting and Filing PMAs” and are intended to be answered by the lead reviewer as an initial screening of the application. FDA does not intend for the applicant to have addressed these items in their application. Depending upon the answers to these preliminary questions, the remainder of the acceptance and filing reviews may or may not be necessary. If the lead reviewer’s responses to the preliminary questions and subsequent consultation with the Center personnel identified below indicate that the PMA acceptance and filing reviews should not continue, the lead reviewer should promptly:
- inform the PMA review team (including consulting reviewers); and
- notify the applicant using proper administrative procedures.
The preliminary questions are:
1. Is the product a device (per section 201(h) of the FD&C Act) or a combination product (per 21 CFR 3.2(e)) with a device constituent part subject to review under PMA?
If the product does not appear to meet the definition of a device under section 201(h) of the FD&CAct, or does not appear to be a combination product with a device constituent part subject to review under PMA, then the lead reviewer should consult with the CDRH Product Jurisdiction Officer or the CBER Product Jurisdiction Officer to determine the appropriate action, and inform division management. If they agree that the product does not appear to be a device or a combination product with a device constituent part subject to review under PMA, the PMA review team should stop the review and notify the applicant.
2. Is the application with the appropriate Center?
If the application is for a single-entity device and appears to be subject to review in a Center different from the one to which it was submitted, or if it is for a combination product with a device constituent part and it appears that a Center different from the one to which it was submitted has the lead, the lead reviewer should consult with the CDRH Product Jurisdiction Officer or the CBER Product Jurisdiction Officer to determine the appropriate action and inform division management. If the PMA is submitted to CDRH and CDRH staff determines that the application is not subject to CDRH review, or the PMA is submitted to CBER and CBER staff determines that the application is not subject to CBER review, the PMA review team should stop the review and notify the applicant electronically.
3.If a Request for Designation (RFD) was submitted for the device or combination product with a device constituent part and assigned to your Center, identify the RFD # and confirm the following:
- Is the device or combination product the same (e.g., design, formulation) as that presented in the RFD submission?
- Are the indications for use for the device or combination product identified in the PMA the same as those identified in the RFD submission?
An RFD determination is specific to the device or combination product and indications for use for the device or combination product described in the RFD submission. If the device or combination product has been modified or the indications for use have been modified since the RFD, the RFD determination may no longer be applicable and jurisdiction may need to be reevaluated by the Office of Combination Products (OCP). The lead reviewer should consult with the CDRH Product Jurisdiction Officer or the CBER Product Jurisdiction Officer to determine the appropriate action and inform division management.
4. Is the applicationfor a combination product that contains as a constituent part a drug that has the same active moiety as an approved drug with exclusivity as described in 21 USC 503(g)(5)(C)(ii)-(v) (section 503(g)(5)(C)(ii)-(v) of the FD&C Act)?
If the application is for a combination product and contains as a constituent a drug that has the same active moiety as an approved drug with exclusivity as described in 21 USC 503(g)(5)(C)(ii)-(v), the lead reviewer should contact the CDRH Product Jurisdiction Officer or CBER Product Jurisdiction Officer to determine the appropriate action and inform division management.
5. Is class III/PMA review required for the device?
Our goal is to apply the appropriate level of regulation to provide a reasonable assurance of safety and effectiveness. Therefore, early in the filing review process, FDA should consider the regulatory burden and the available mechanisms to apply the proper degree of regulation. In making this determination, staff should consider how similar devices are being regulated.
Class III devices are those that cannot be classified as Class I or Class II devices because insufficient information exists to determine that general and special controls are sufficient to provide reasonable assurance of the safety and effectiveness of the device, and either (1) are purported to be for a use in supporting or sustaining human life or for a use which is of substantial importance in preventing impairment of human health; or (2) present a potential unreasonable risk of illness or injury. See section 513(a)(1)(C) of the FD&CAct. Devices may also automatically be classified in class III under section 513(f)(1) of the FD&CAct.
Generally, PMA review is required if the device is:
- a transitional device that has not been reclassified (see section 520(l) of the FD&C Act),
- the subject of a final “call for PMA” under section 515(b) of the FD&CAct, or
- automatically classified into Class III under section 513(f) of the FD&CAct, including devices found to be Not Substantially Equivalent (NSE) in response to a 510(k) premarket notification and/or for which a request for reclassification under 513(f)(2) has been denied by FDA.
If regulation under PMA does not appear to be required, the PMA lead reviewer should consult division management and other Center resources to determine the appropriate action. If the review division agrees that review in a different type of marketing application may be an option, the PMA review team should notify the applicant to discuss the most appropriate path forward.
6. Is there a pending 510(k) for the same device with the same indications for use?
FDA may decide not to file a PMA if the applicant has a 510(k) for the same device with the same indications for use pending (21 CFR 814.42(e)(3)). If there is a pending 510(k), the review team should stop the review. The lead reviewer should consult division management and other Center resources to determine which premarket review pathway applies to the device and the appropriate processes for addressing the situation. Staff should also consult division management and other Center resources if a 510(k) and PMA have been submitted for the same device type by different applicants.
7. Is the applicant the subject of the Application Integrity Policy (AIP)14?
The lead reviewer should refer to the AIP list.15 If the applicant is on the list, the reviewer should consult the CDRH Office of Product Evaluation and Quality/Office of Clinical Evidence and Analysis/Division of Clinical Science and Quality (OPEQ/OCEA/DCEA1) or CBER Office of Compliance and Biologics Quality/Division of Inspections and Surveillance/Bioresearch Monitoring Branch (OCBQ/DIS/BMB) to determine the appropriate action.
IX. The Checklist – Acceptance Review 🔗
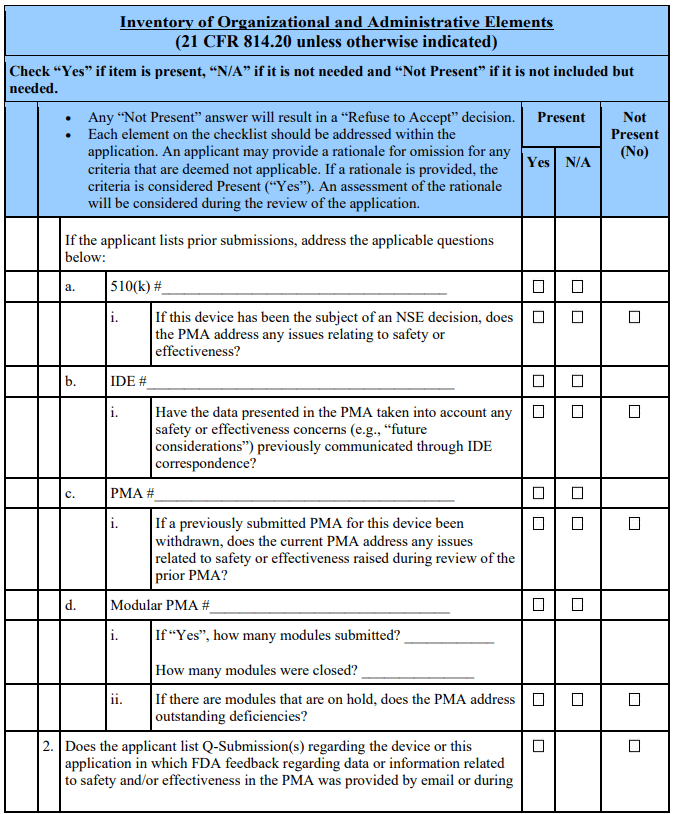
If the answers to the above preliminary questions indicate that PMA review should continue, the acceptance review should proceed by answering questions in the “Acceptance Review” section of the checklist. This section of the checklist collects information regarding the completeness of the PMA (i.e., “Inventory of Organizational and Administrative Elements”) and guides FDA staff through the process necessary to arrive at a decision to “Accept” a PMA or designate it “Refuse to Accept.”
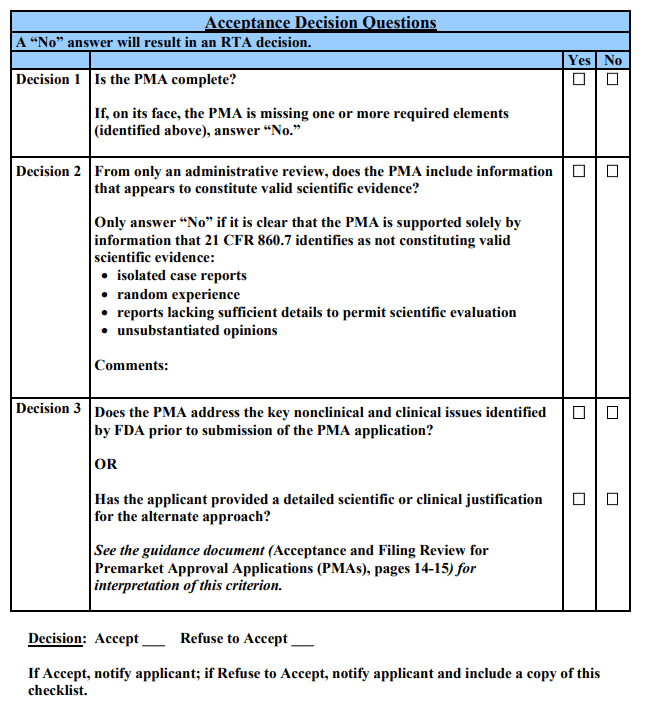
The specific issues that are critical to the PMA acceptance decision-making process (i.e., the “Acceptance Decision Questions”) are individually discussed below. The numbering scheme used for these decision questions corresponds to the checklist. Each Acceptance Decision Question should be answered. Only if all questions are answered “Yes” can the PMA application be accepted for filing review.
Acceptance Decision 1: Is the PMA administratively complete?
The questions in Section A of the checklist are intended to outline each of the
administrative elements required by 21 CFR 814.20 that are necessary for substantive review of the PMA. If, on its face, the PMA is missing one or more required elements or sections as described by the questions in Section A (including manufacturing information as discussed above), the answer to the above question is “No” and the lead reviewer should note the specific omission(s) on the checklist. A section will be considered missing if it is not in English or not accompanied by an English translation. If such omissions exist, the FDA should not accept the PMA.
Acceptance Decision 2: From only an administrative review, does the PMA include data that appears to constitute valid scientific evidence?
The answer to this question is “No” if it is clear that the only information provided in the PMA is information that is not regarded as valid scientific evidence under 21 CFR 860.7 (i.e., “isolated case reports, random experience, reports lacking sufficient details to permit scientific evaluation, and unsubstantiated opinions”). If none of the data, on their face, constitute valid scientific evidence, the PMA should not be accepted.
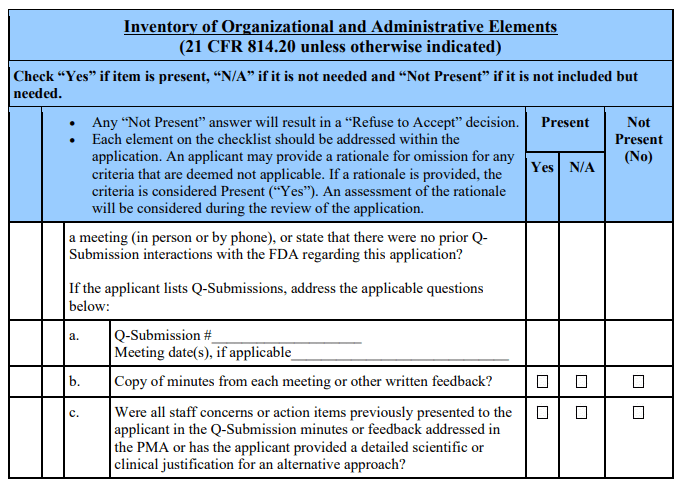
Acceptance Decision 3: Does the PMA address the key nonclinical and clinical issues identified by FDA prior to submission of the PMA application, OR has the applicant provided a scientific or clinical justification for an alternative approach?
Section B of the checklist outlines questions intended to identify when the FDA has previously provided specific guidance to the applicant about the content of the PMA through one or more mechanisms, such as a prior PMA application, a prior “Not Substantially Equivalent” decision on a 510(k), Investigational Device Exemption (IDE) letters, Q-Submission feedback, a Determination or Agreement meeting(s), or other substantive communication with FDA, or through a published guidance document. If such information has been communicated to the applicant through one or more of these mechanisms, and the PMA application addresses each of the key nonclinical and clinical issues identified by FDA, the answer to the above question is “Yes.” Furthermore, if some of these key issues previously identified by FDA are not addressed, but the PMA application contains a scientific or clinical justification for the omission or deviation, the answer to the above question is “Yes.” If the PMA application contains a statement that there were no prior submissions for the device, the answer to the above question is “Yes.” These cases do not preclude the review division from accepting the PMA.
In this context, the term “key issues” is meant to refer to issues that are central to our review of device safety and effectiveness under section 515(c) and (d) of the FD&CAct. Examples of key issues include: need for long-term nonclinical studies (e.g., biocompatibility, carcinogenicity, or other animal studies), and certain clinical trial parameters (e.g., sample size, patient population, statistical hypothesis, study design, and endpoints). These key issues typically are device-specific. As a result, the decision of the review division to “Refuse to Accept” a PMA application based on this criterion can only be made after carefully considering the following questions:
Are the types of necessary nonclinical and clinical studies well-known in the scientific and medical communities for the particular device?
For an “established” device type, the types of nonclinical and clinical studies that we would expect in a PMA are likely to be well-known both within FDA and in the scientific and medical communities and, as such, are often included as part of an FDA guidance
document and/or consensus standard. If citing voluntary consensus standards, the applicant should consider FDA’s guidance “Appropriate Use of Voluntary Consensus Standards in Premarket Submissions for Medical Devices.”16
Were the issues conveyed to the applicant as part of a documented regulatory process?
Examples of a documented regulatory process include:
- Q-submission interaction,
- prior PMA application,
- prior “Not Substantially Equivalent” decision on a 510(k),
- IDE letters, or
- letter(s) issued as a result of Determination or Agreement meetings.
Staff should only designate a PMA “Refuse to Accept” based on a “No” response to “Acceptance Decision 3” in instances where the key issues were identified by staff as part of a documented regulatory process.
X. The Checklist – Filing Review 🔗
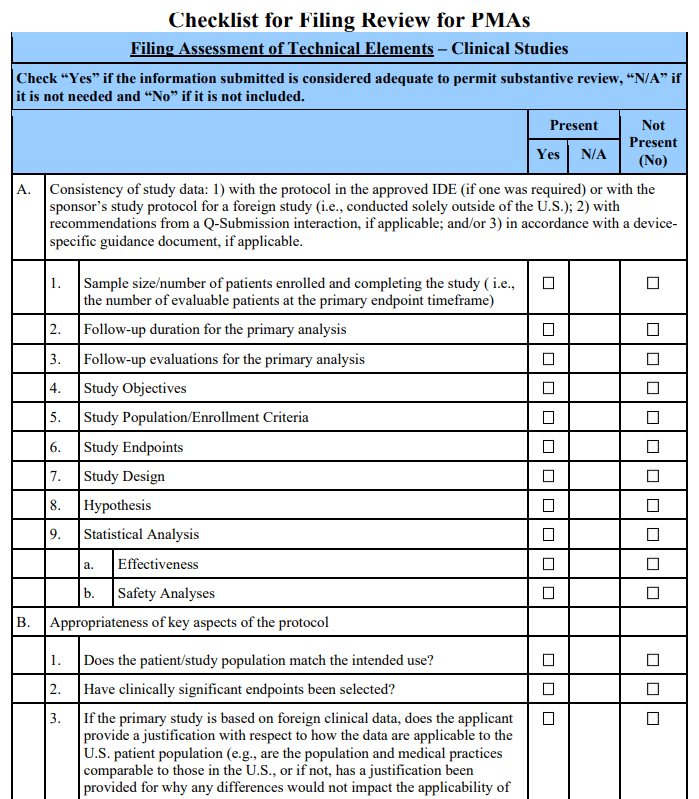
If the answers to the above preliminary questions and acceptance decision questions indicate that PMA review should continue, the formal filing review should proceed by answering questions in the “Filing Review” section of the checklist. This section of the checklist assesses the basic adequacy of the technical elements (i.e., “Filing Assessment of Technical Elements”) and guides FDA staff through the process necessary to arrive at a decision to “File” or “Not File” a PMA.
The specific issues that are critical to the PMA filing decision-making process (i.e., the “Filing Decision Questions”) are individually discussed below. The numbering scheme used for these decision questions corresponds to that of the checklist. Each Filing Decision Question should be answered. Only if all questions are answered “Yes” can the PMA application be filed.
We do not anticipate that a single member of the PMA review team will be able to answer all of these questions. Rather, we expect that the lead reviewer will complete this checklist in consultation with the team members, in particular the medical officer and statistician.
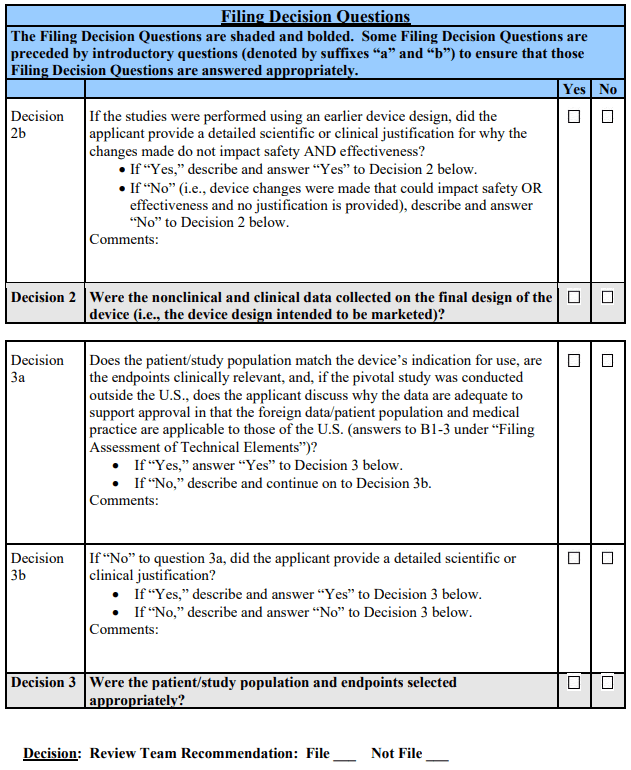
Filing Decision 1: Were the clinical study data collected and analyzed per the protocol?
If the clinical data submitted in support of PMA approval appear, on an administrative review, to have been collected and analyzed consistent with the major elements of the clinical protocol (i.e., objectives, study population, endpoints, study design, hypothesis, sample size, and follow-up duration), or the applicant provides a scientific or clinical justification for the use of an alternative approach, the answer to the above question is “Yes” and the lead reviewer will note any specific deviations or justifications on the checklist. In addition, if the sample size is smaller or the follow-up duration is shorter than specified in the clinical protocol, but such changes are supported by either: (i) the recommendation of a Data Monitoring Committee (DMC) or (ii) statistical plans that incorporate interim stopping rules, substantive review of the PMA may proceed. That is, these cases do not preclude the FDA from filing the PMA.
If the study deviated from the clinical protocol with respect to the major elements identified in the paragraph above and the applicant provided no justification for doing so, the answer to the above question is “No” and the lead reviewer will note the specific deviation(s) on the checklist. In these cases, the FDA should not file the PMA.
As discussed above, occasionally, applicants have submitted PMAs with incomplete clinical data (i.e., the sample size is smaller or follow-up duration for the primary analysis is shorter than specified in the clinical protocol). If no justification is provided and/or the applicant indicates they intend to update the PMA with necessary additional clinical data, we will consider such PMAs to be submitted prematurely and therefore incomplete. If the PMA is viewed as a premature application, the answer to the above question is “No.” In these cases, the FDA should not file the PMA.
Filing Decision 2: Were the nonclinical and clinical data collected on the final design of the device (i.e., the device design intended to be marketed)?
If the nonclinical and pivotal clinical data submitted in support of PMA approval were collected on the final device design, or the differences between the study device and final device clearly do not affect safety or effectiveness of the device and/or clinical outcome, the answer to the above question is “Yes” and any device changes will be noted on the checklist. Furthermore, if the clinical data were collected on an earlier design of the device and the applicant provides a scientific or clinical justification describing why the study results on the earlier device design apply to the proposed design, the answer to the above question is “Yes” and the justification will be noted on the checklist. These cases do not preclude the FDA from filing the PMA.
If changes that could potentially impact safety and/or effectiveness were made to the device design either during or after the pivotal nonclinical and clinical studies, and no justification is provided as to why these data are applicable to the new design, the answer to the above question is “No.” In this case, the lead reviewer will note the specific device change(s) on the checklist, and the FDA should not file the PMA.
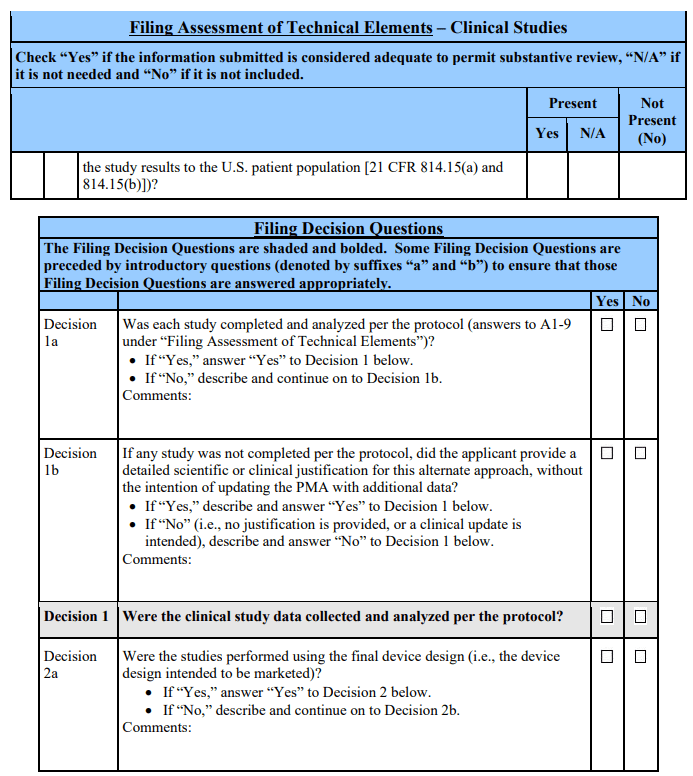
Filing Decision 3: Were the patient/study17 population and endpoints consistent with the proposed indications?
If, upon an administrative review, the patient population (as defined by the inclusion and exclusion criteria) in the pivotal study appears to match the device’s proposed indications for use and the endpoints that were selected were agreed to by FDA and/or appear to be clinically relevant, the answer to the above question is “Yes.” Additionally, if the patient population and/or endpoints are inconsistent with the proposed indications but the applicant provides a detailed scientific or clinical justification for this approach, the answer to the above question is “Yes.” These cases do not preclude the FDA from filing the PMA.
If either the patient population or endpoints of the pivotal study, on their face, do not match the proposed indications for use and no justification is provided for this alternative approach, the answer to the above question is “No.” In addition, if the pivotal study was conducted outside the U.S. and the applicant has not addressed how such data are adequate to support approval (including addressing how the local medical practice and/or patient population match those of the U.S. or why any differences would not impact the applicability of the study results to the U.S. patient population), answer “No” to the above question. In these cases, the PMA should not be filed.
Appendix A. Checklists for Acceptance and Filing of PMAs 🔗


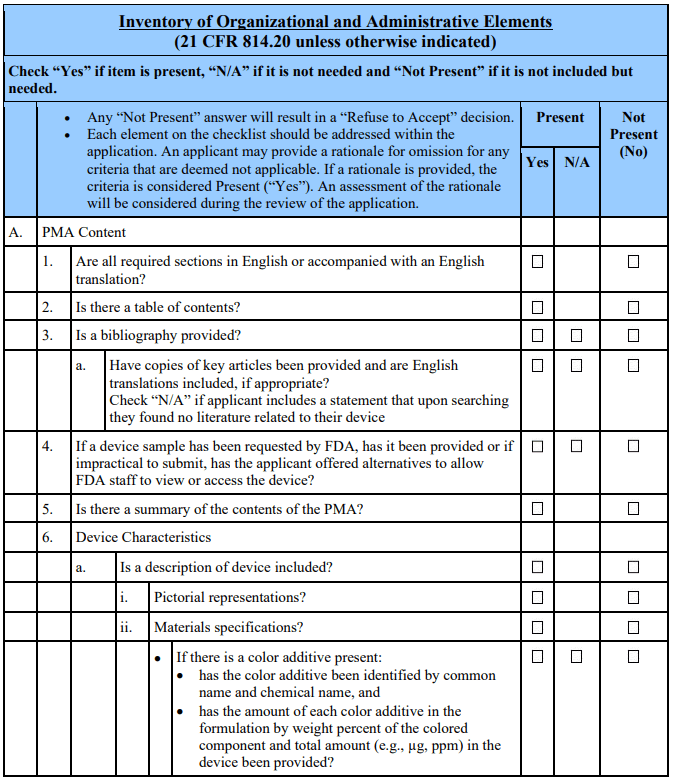
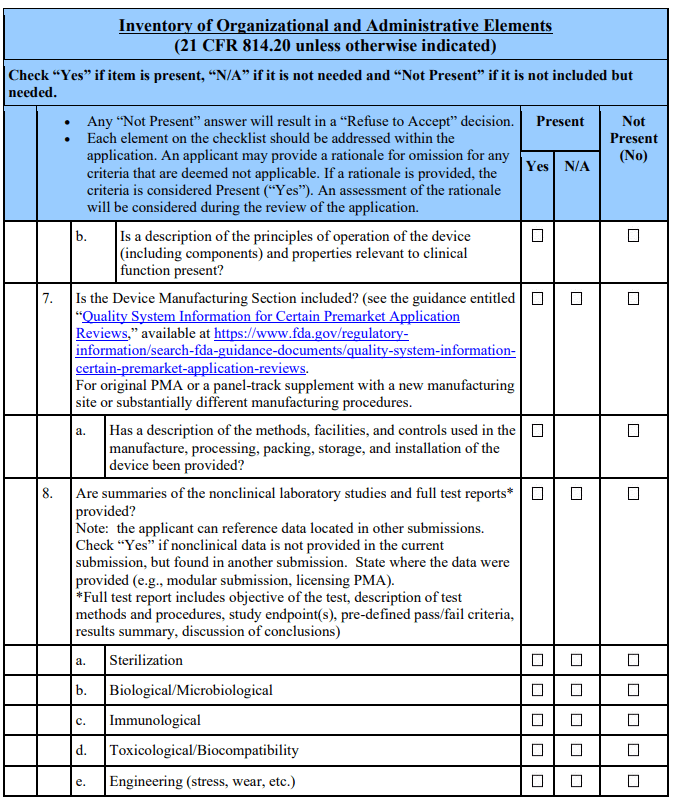
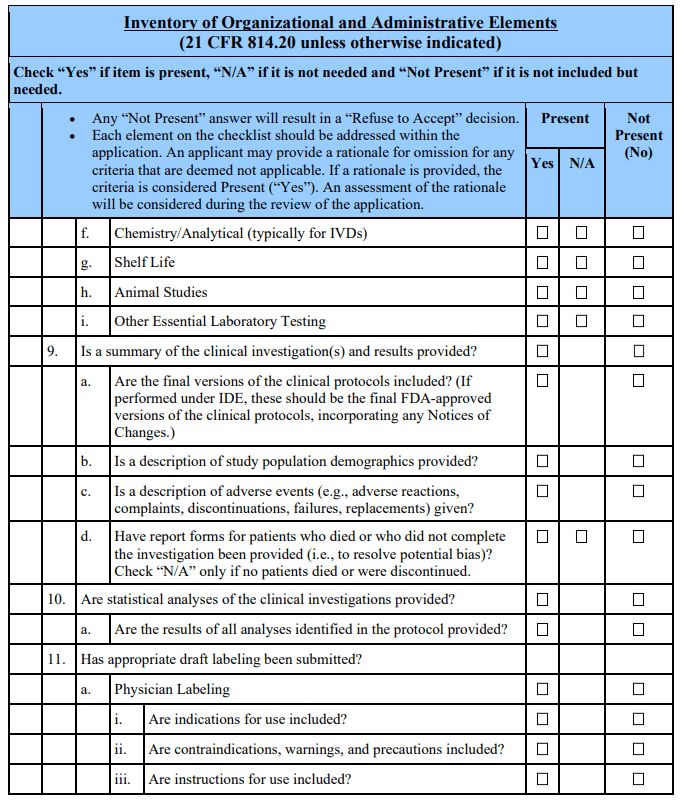
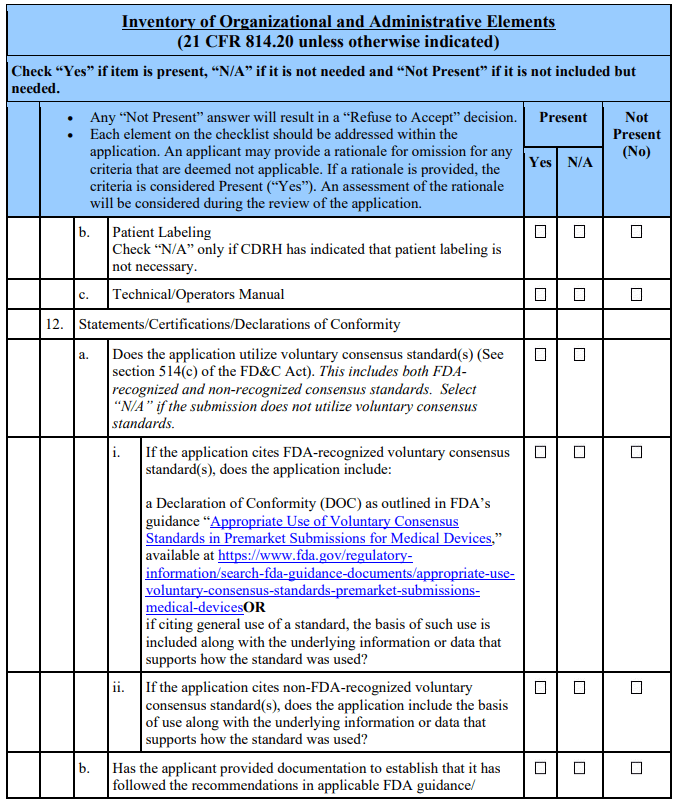

Footnotes 🔗
-
See Title II of the FDA Reauthorization Act of 2017 (Public Law 115-52) ↩
-
There are three (3) criteria for not processing a PMA that has been received: i) the application is not submitted with the required user fee and user fee payment identification (ID) number (provided on the MDUFA User Fee Cover Sheet (Form 3601), the application cover letter, or the CDRH Premarket Review Submission Cover Sheet (Form 3514) per the Medical Device User Fee Amendments of 2012, ii) the application is not signed or countersigned by a U.S. representative per 21 CFR 814.20(a), and iii) the firm did not submit the correct number of copies per 814.20(b)(2). Since any PMA not meeting these three criteria will not be processed by the CDRH Document Mail Center or CBER Regulatory Project Manager, they are not included in the checklist. ↩
-
See the guidance “eCopy Program for Medical Device Submissions,” available at https://www.fda.gov/regulatoryinformation/search-fda-guidance-documents/ecopy-program-medical-device-submissions ↩
-
An acceptance and filing review will be conducted on modular PMAs at the time of receipt of the final clinical module, which completes the PMA application, and will be an assessment of all information in all modules received to date. Please refer to the guidance document entitled “Premarket Approval Application Modular Review,” available at https://www.fda.gov/regulatory-information/search-fda-guidance-documents/premarket-approvalapplication-modular-, for additional information regarding Modular PMAs. ↩
-
See section 515B of the FD&C Act ↩
-
Available at https://www.fda.gov/regulatory-information/search-fda-guidance-documents/breakthrough-devicesprogram. ↩
-
Available at https://www.fda.gov/medical-devices/premarket-submissions/medical-device-user-fees ↩
-
Available at https://www.fda.gov/regulatory-information/search-fda-guidance-documents/requests-feedback-andmeetings-medical-device-submissions-q-submission-program. ↩
-
Available at https://www.fda.gov/medical-devices/device-advice-comprehensive-regulatory-assistance. ↩
-
Available at https://www.fda.gov/medical-devices/device-advice-comprehensive-regulatory-assistance/guidancedocuments-medical-devices-and-radiation-emitting- ↩
-
Available at https://www.fda.gov/regulatory-information/search-fda-guidance-documents/modifications-devices-subject-premarket-approval-pma-pma-supplement-decision-making-process ↩
-
For additional information, please see the guidance “FDA and Industry Actions on Premarket Approval Applications (PMAs): Effect on FDA Review Clock and Goals” available at https://www.fda.gov/regulatory-information/search-fda-guidance-documents/fda-and-industry-actions-premarket-approval-applications-pmas-effect-fda-review-clock-and-. ↩
-
3See 163 CONG. REC. S4729-S4736 (daily ed. August 2, 2017) (Food and Drug Administration User Fee
Reauthorization), also available at https://www.fda.gov/media/102699/download. ↩ -
When data in a pending application has been called into question by certain wrongful acts (fraud, untrue statements of material facts, bribery, or illegal gratuities), FDA intends to defer substantive scientific review of such data until completion of a validity assessment and questions regarding reliability of the data are resolved. (See FDA Guide 7150.09 Compliance Policy Guide, Chapter 50 – General Policy – Subject: Fraud, Untrue Statements of Material Facts, Bribery, and Illegal Gratuities, 56 FR 46191). ↩
-
Available at https://www.fda.gov/inspections-compliance-enforcement-and-criminal-investigations/application-integrity-policy/application-integrity-policy-list. ↩
-
Available at https://www.fda.gov/regulatory-information/search-fda-guidance-documents/appropriate-use-voluntary-consensus-standards-premarket-submissions-medical-devices. ↩
-
Note that in the case of PMAs submitted to CBER, the study population may be blood donors rather than patients. ↩
