
Innolitics Introduction 🔗
This document contains the current human factors guidance (as of June 2023). There is a 2022 draft guidance that will complement this once it’s finalized.
About this Transcript 🔗
This document is a transcript of an official FDA (or IMDRF) guidance document. We transcribe the official PDFs into HTML so that we can share links to particular sections of the guidance when communicating internally and with our clients. We do our best to be accurate and have a thorough review process, but occasionally mistakes slip through. If you notice a typo, please email a screenshot of it to Mihajlo at mgrcic@innolitics.com so we can fix it.
Preamble 🔗
Document issued on February 3, 2016.
For questions regarding this document, contact the Human Factors Premarket Evaluation Team at (301) 796-5580.
1. Introduction 🔗
FDA has developed this guidance document to assist industry in following appropriate human factors and usability engineering processes to maximize the likelihood that new medical devices will be safe and effective for the intended users, uses and use environments.
The recommendations in this guidance document are intended to support manufacturers in improving the design of devices to minimize potential use errors and resulting harm. The FDA believes that these recommendations will enable manufacturers to assess and reduce risks associated with medical device use.
FDA's guidance documents, including this one, do not establish legally enforceable responsibilities. Instead, guidance documents describe the Agency's current thinking on a topic and should be viewed only as recommendations unless specific regulatory or statutory requirements are cited. The use of the word should in Agency guidance documents means that something is suggested or recommended, but not required.
2. Scope 🔗
This guidance recommends that manufacturers follow human factors or usability engineering processes during the development of new medical devices, focusing specifically on the user interface, where the user interface includes all points of interaction between the product and the user(s) including elements such as displays, controls, packaging, product labels, instructions for use, etc. While following these processes can be beneficial for optimizing user interfaces in other respects (e.g., maximizing ease of use, efficiency, and user satisfaction), FDA is primarily concerned that devices are safe and effective for the intended users, uses, and use environments. The goal is to ensure that the device user interface has been designed such that use errors that occur during use of the device that could cause harm or degrade medical treatment are either eliminated or reduced to the extent possible.
As part of their design controls1, manufacturers conduct a risk analysis that includes the risks associated with device use and the measures implemented to reduce those risks. ANSI/AAMI/ISO 14971, Medical Devices – Application of risk management to medical devices, defines risk as the combination of the probability of occurrence of harm and the severity of the potential harm2. However, because probability is very difficult to determine for use errors, and in fact many use errors cannot be anticipated until device use is simulated and observed, the severity of the potential harm is more meaningful for determining the need to eliminate (design out) or reduce resulting harm. If the results of risk analysis indicate that use errors could cause serious harm to the patient or the device user, then the manufacturer should apply appropriate human factors or usability engineering processes according to this guidance document. This is also the case if a manufacturer is modifying a marketed device to correct design deficiencies associated with use, particularly as a corrective and preventive action (CAPA).
CDRH considers human factors testing a valuable component of product development for medical devices. CDRH recommends that manufacturers consider human factors testing for medical devices as a part of a robust design control subsystem. CDRH believes that for those devices where an analysis of risk indicates that users performing tasks incorrectly or failing to perform tasks could result in serious harm, manufacturers should submit human factors data in premarket submissions (i.e., PMA, 510(k)). In an effort to make CDRH’s premarket submission expectations clear regarding which device types should include human factors data in premarket submissions, CDRH is issuing a draft guidance document List of Highest Priority Devices for Human Factors Review, Draft Guidance for Industry and Food and Drug Administration Staff. (http://www.fda.gov/downloads/MedicalDevices/DeviceRegulationandGuidance/GuidanceDocuments/UCM484097.pdf) When final, this document will represent the Agency’s current thinking on this issue.
3. Definitions 🔗
For the purposes of this guidance, the following terms are defined.
3.1 Abnormal use 🔗
An intentional act or intentional omission of an act that reflects violative or reckless use or sabotage beyond reasonable means of risk mitigation or control through design of the user interface.
3.2 Critical task 🔗
A user task which, if performed incorrectly or not performed at all, would or could cause serious harm to the patient or user, where harm is defined to include compromised medical care.
3.3 Formative evaluation 🔗
Process of assessing, at one or more stages during the device development process, a user interface or user interactions with the user interface to identify the interface’s strengths and weaknesses and to identify potential use errors that would or could result in harm to the patient or user.
3.4 Hazard 🔗
Potential source of harm.
3.5 Hazardous situation 🔗
Circumstance in which people are exposed to one or more hazard(s).
3.6 Human factors engineering 🔗
The application of knowledge about human behavior, abilities, limitations, and other characteristics of medical device users to the design of medical devices including mechanical and software driven user interfaces, systems, tasks, user documentation, and user training to enhance and demonstrate safe and effective use. Human factors engineering and usability engineering can be considered to be synonymous.
3.7 Human factors validation testing 🔗
Testing conducted at the end of the device development process to assess user interactions with a device user interface to identify use errors that would or could result in serious harm to the patient or user. Human factors validation testing is also used to assess the effectiveness of risk management measures. Human factors validation testing represents one portion of design validation.
3.8 Task 🔗
Action or set of actions performed by a user to achieve a specific goal.
3.9 Use error 🔗
User action or lack of action that was different from that expected by the manufacturer and caused a result that (1) was different from the result expected by the user and (2) was not caused solely by device failure and (3) did or could result in harm.
3.10 Use safety 🔗
Freedom from unacceptable use-related risk.
3.11 User 🔗
Person who interacts with (i.e., operates or handles) the device.
3.12 User interface 🔗
All points of interaction between the user and the device, including all elements of the device with which the user interacts (i.e., those parts of the device that users see, hear, touch). All sources of information transmitted by the device (including packaging, labeling), training and all physical controls and display elements (including alarms and the logic of operation of each device component and of the user interface system as a whole).
4. Overview 🔗
Understanding how people interact with technology and studying how user interface design affects the interactions people have with technology is the focus of human factors engineering (HFE) and usability engineering (UE)3.
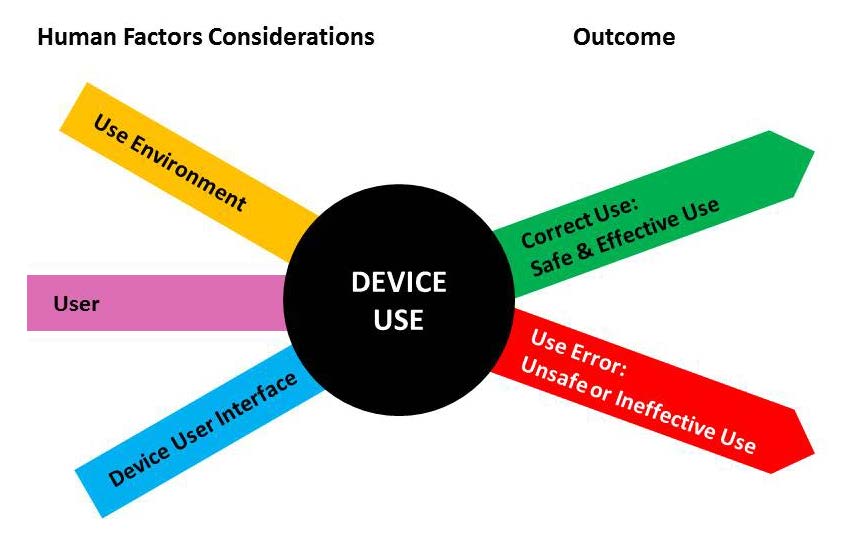
HFE/UE considerations in the development of medical devices involve the three major components of the device-user system: (1) device users, (2) device use environments and (3) device user interfaces. The interactions among the three components and the possible results are depicted graphically in Figure 1.
4.1 HFE/UE as Part of Risk Management 🔗
Eliminating or reducing design-related problems that contribute to or cause unsafe or ineffective use is part of the overall risk management process.
Hazards traditionally considered in risk analysis include:
- Physical hazards (e.g., sharp corners or edges),
- Mechanical hazards (e.g., kinetic or potential energy from a moving object),
- Thermal hazards (e.g., high-temperature components),
- Electrical hazards (e.g., electrical current, electromagnetic interference (EMI)),
- Chemical hazards (e.g., toxic chemicals),
- Radiation hazards (e.g., ionizing and non-ionizing), and
- Biological hazards (e.g., allergens, bio-incompatible agents and infectious agents).
These hazards are generally associated with instances of device or component failure that are not dependent on how the user interacts with the device. (A notable exception is infectious agents (germs/pathogens), which can be introduced to the device as cross contamination caused by use error.)
Medical device hazards associated with user interactions with devices should also be included in risk management. These hazards are referred to in this document as use-related hazards (see Figure 2). These hazards might result from aspects of the user interface design that cause the user to fail to adequately or correctly perceive, read, interpret, understand or act on information from the device. Some use-related hazards are more serious than others, depending on the severity of the potential harm to the user or patient encountering the hazard.
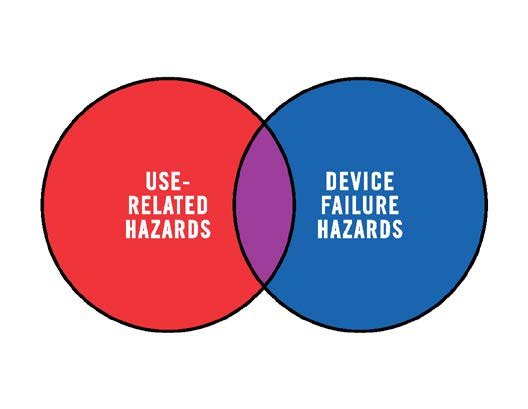
Use-related hazards are related to one or more of the following situations:
- Device use requires physical, perceptual, or cognitive abilities that exceed the abilities of the user;
- Device use is inconsistent with the user’s expectations or intuition about device operation;
- The use environment affects operation of the device and this effect is not recognized or understood by the user;
- The particular use environment impairs the user’s physical, perceptual, or cognitive capabilities when using the device;
- Devices are used in ways that the manufacturer could have anticipated but did not consider; or
- Devices are used in ways that were anticipated but inappropriate (e.g., inappropriate user habits) and for which risk elimination or reduction could have been applied but was not.
4.2 Risk Management 🔗
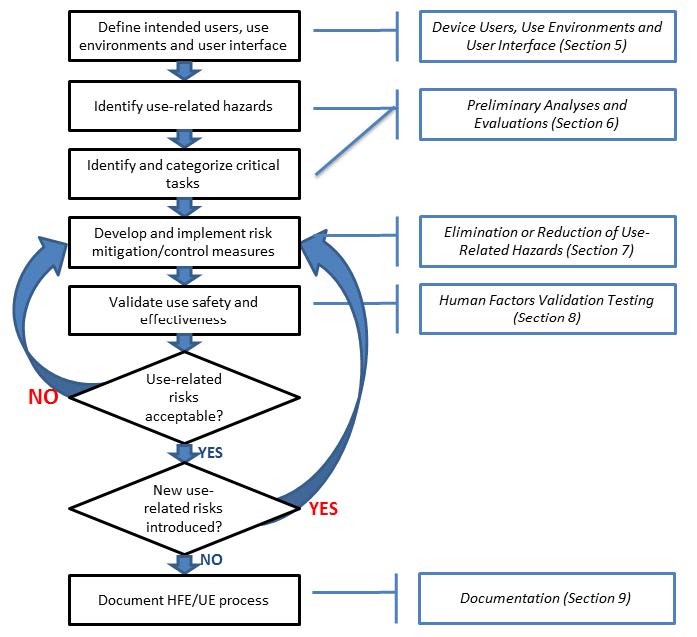
HFE/UE considerations and approaches should be incorporated into device design, development and risk management processes. Three steps are essential for performing a successful HFE/UE analysis:
- Identify anticipated use-related hazards and initially unanticipated use-related hazards (derived through preliminary analyses and evaluations, see Section 6), and determine how hazardous use situations occur;
- Develop and apply measures to eliminate or reduce use-related hazards that could result in harm to the patient or the user (see Section 7); and
- Demonstrate whether the final device user interface design supports safe and effective use by conducting human factors validation testing (see Section 8).
Figure 3 depicts the risk management process for addressing use-related hazards; HFE/UE approaches should be applied for this process to work effectively.
5. Device Users, Use Environments and User Interface 🔗
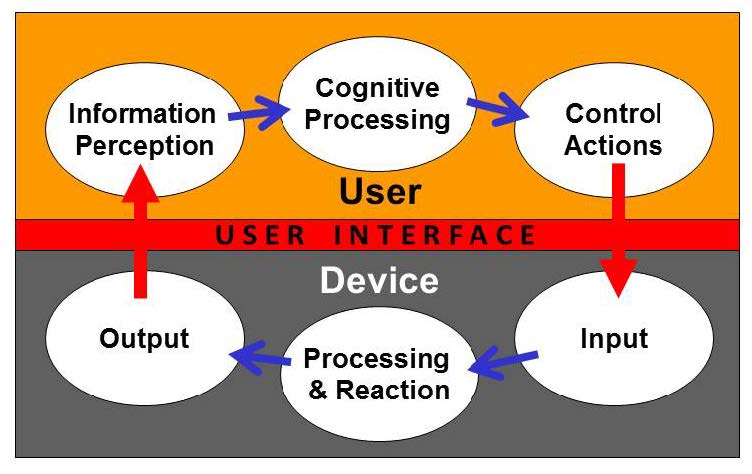
Figure 4 presents a model of the interactions between a user and a device, the processes performed by each, and the user interface between them. When users interact with a device, they perceive information provided by the device, then interpret and process the information and make decisions. The users interact with the device to change some aspect of its operation (e.g., modify a setting, replace a component, or stop the device). The device receives the user input, responds, and provides feedback to the user. The user might then consider the feedback and initiate additional cycles of interaction.
Prior to conducting HFE/UE analyses you should review and document essential characteristics of the following:
- Device users; e.g.:
- The intended users of the device (e.g., physician, nurse, professional caregiver, patient, family member, installer, maintenance staff member, reprocessor, disposer);
- User characteristics (e.g., functional capabilities (physical, sensory and cognitive), experience and knowledge levels and behaviors) that could impact the safe and effective use of the device; and
- The level of training users are expected to have and/or receive.
- Device use environments; e.g.:
- Hospital, surgical suite, home, emergency use, public use, etc.; or
- Special environments (e.g., emergency transport, mass casualty event, sterile isolation, hospital intensive care unit).
- Device user interface; e.g.:
- Components and accessories
- Controls
- Visual displays
- Visual, auditory and tactile feedback
- Alarms and alerts
- Logic and sequence of operation
- Labeling
- Training
These considerations are discussed in more detail in the following sections. The characteristics of the intended users, use environments, and the device user interface should be taken into account during the medical device development process.
5.1 Device Users 🔗
The intended users of a medical device should be able to use it without making use errors that could compromise medical care or patient or user safety. Depending on the specific device and its application, device users might be limited to professional caregivers, such as physicians, nurses, nurse practitioners, physical and occupational therapists, social workers, and home care aides. Other user populations could include medical technologists, radiology technologists, or laboratory professionals. Device user populations might also include the professionals who install and set up the devices and those who clean, maintain, repair, or reprocess them. The users of some devices might instead be non-professionals, including patients who operate devices on themselves to provide self-care and family members or friends who serve as lay caregivers to people receiving care in the home, including parents who use devices on their children or supervise their children’s use of devices.
The ability of a user to operate a medical device depends on his or her personal characteristics, including:
- Physical size, strength, and stamina,
- Physical dexterity, flexibility, and coordination,
- Sensory abilities (i.e., vision, hearing, tactile sensitivity),
- Cognitive abilities, including memory,
- Medical condition for which the device is being used,
- Comorbidities (i.e., multiple conditions or diseases),
- Literacy and language skills,
- General health status,
- Mental and emotional state,
- Level of education and health literacy relative to the medical condition involved,
- General knowledge of similar types of devices,
- Knowledge of and experience with the particular device,
- Ability to learn and adapt to a new device, and
- Willingness and motivation to learn to use a new device.
You should evaluate and understand the characteristics of all intended user groups that could affect their interactions with the device and describe them for the purpose of HFE/UE evaluation and design. These characteristics should be taken into account during the medical device development process, so that devices might be more accommodating of the variability and limitations among users.
5.2 Device Use Environments 🔗
The environments in which medical devices are used might include a variety of conditions that could determine optimal user interface design. Medical devices might be used in clinical environments or non-clinical environments, community settings or moving vehicles. Examples of environmental use conditions include the following:
- The lighting level might be low or high, making it hard to see device displays or controls.
- The noise level might be high, making it hard to hear device operation feedback or audible alerts and alarms or to distinguish one alarm from another.
- The room could contain multiple models of the same device, component or accessory, making it difficult to identify and select the correct one.
- The room might be full of equipment or clutter or busy with other people and activities, making it difficult for people to maneuver in the space and providing distractions that could confuse or overwhelm the device user.
- The device might be used in a moving vehicle, subjecting the device and the user to jostling and vibration that could make it difficult for the user to read a display or perform fine motor movements.
You should evaluate and understand relevant characteristics of all intended use environments and describe them for the purpose of HFE/UE evaluation and design. These characteristics should be taken into account during the medical device development process, so that devices might be more accommodating of the conditions of use that could affect their use safety and effectiveness.
5.3 Device User Interface 🔗
A device user interface includes all points of interaction between the user and the device, including all elements of the device with which the user interacts. A device user interface might be used while user setups the device (e.g., unpacking, set up, calibration), uses the device, or performs maintenance on the device (e.g., cleaning, replacing a battery, repairing parts).
It includes:
- The size and shape of the device (particularly a concern for hand-held and wearable devices),
- Elements that provide information to the user, such as indicator lights, displays, auditory and visual alarms,
- Graphic user interfaces of device software systems,
- The logic of overall user-system interaction, including how, when, and in what form information (i.e., feedback) is provided to the user,
- Components that the operator connects, positions, configures or manipulates,
- Hardware components the user handles to control device operation such as switches, buttons, and knobs,
- Components or accessories that are applied or connected to the patient, and
- Packaging and labeling, including operating instructions, training materials, and other materials.
The most effective strategies to employ during device design to reduce or eliminate use-related hazards involve modifications to the device user interface. To the extent possible, the “look and feel” of the user interface should be logical and intuitive to use. A well-designed user interface will facilitate correct user actions and will prevent or discourage actions that could result in harm (use errors). Addressing use-related hazards by modifying the device design is usually more effective than revising the labeling or training. In addition, labeling might not be accessible when needed and training depends on memory, which might not be accurate or complete.
An important aspect of the user interface design is the extent to which the logic of information display and control actions is consistent with users’ expectations, abilities, and likely behaviors at any point during use. Users will expect devices and device components to operate in ways that are consistent with their experiences with similar devices or user interface elements. For example, users might expect the flow rate of a liquid or gaseous substance to increase or to decrease by turning a control knob in a specific direction based on their previous experiences. The potential for use error increases when this expectation is violated, for example, when an electronically-driven control dial is designed to be turned in the opposite direction of dials that were previously mechanical.
Increasingly, user interfaces for new medical devices are software-driven. In these cases, the user interface might include controls such as a keyboard, mouse, stylus, touchscreen; future devices might be controlled through other means, such as by gesture, eye gaze, or voice. Other features of the user interface include the manner in which data is organized and presented to users. Displayed information typically has some form of hierarchical structure and navigation logic.
6. Preliminary Analyses and Evaluations 🔗
Preliminary analyses and evaluations are performed to identify user tasks, user interface components and use issues early in the design process. These analyses help focus the HFE/UE processes on the user interface design as it is being developed so it can be optimized with respect to safe and effective use. One of the most important outcomes of these analyses is comprehensive identification and categorization of user tasks, leading to a list of critical tasks (Section 6.1).
Human factors and usability engineering offer a variety of methods for studying the interactions between devices and their users. Your choice of approaches to take when developing a new or modified device is dependent on many factors related to the specific device development effort, such as the level of novelty of the planned device and your initial level of knowledge of the device type and the device users.
Frequently-used HFE/UE analysis and evaluation methods are discussed below. They can be used to identify problems known to exist with previous versions of the device or device type (Section 6.2). Analytical methods (Section 6.3) and empirical methods (Section 6.4) can be useful for identifying use-related hazards and hazardous situations. These techniques are discussed separately; however, they are interdependent and should be employed in complementary ways. The results of these analyses and evaluations should be used to inform your risk management efforts (Section 7) and development of the protocol for the human factors validation test (Section 8).
6.1 Critical Task Identification and Categorization 🔗
An essential goal of the preliminary analysis and evaluation process is to identify critical tasks that users should perform correctly for use of the medical device to be safe and effective.
You should categorize the user tasks based on the severity of the potential harm that could result from use errors, as identified in the risk analysis. The purpose is to identify the tasks that, if performed incorrectly or not performed at all, would or could cause serious harm. These are the critical tasks. Risk analysis approaches, such as failure modes effects analysis (FMEA) and fault tree analysis (FTA) can be helpful tools for this purpose.
All risks associated with the warnings, cautions and contraindications in the labeling should be included in the risk assessment. Reasonably foreseeable misuse (including device use by unintended but foreseeable users) should be evaluated to the extent possible, and the labeling should include specific warnings describing that use and the potential consequences. Abnormal use is generally not controllable through application of HFE/UE processes.
The list of critical tasks is dynamic and will change as the device design evolves and the preliminary analysis and evaluation process continues. As user interactions with the user interface become better understood, additional critical tasks will likely be identified and be added to the list. The final list of critical tasks is used to structure the human factors validation test to ensure it focuses on the tasks that relate to device use safety and effectiveness. Note that some potential use errors might not be recognized until the human factors validation testing is conducted, which is why the test protocol should include mechanisms to detect previously unanticipated use errors.
6.1.1 Failure mode effects analysis 🔗
Applying a failure mode effects analysis approach to analysis of use safety is most successful when performed by a team consisting of people from relevant specialty areas. The analysis team might include individuals with experience using the device such as a patient who uses the device or a clinical expert and also a design engineer and a human factors specialist. The team approach ensures that the analysis includes multiple viewpoints on potential use errors and the harm that could result. The FMEA team “brainstorms” possible use scenarios that could lead to a “failure mode” and considers the tasks and potential harm for each possible use error.
A task analysis can be helpful in this process by describing user–device interaction. The task analysis should also be refined during the FMEA process.
6.1.2 Fault tree analysis 🔗
Fault tree analysis (FTA) differs from FMEA in that it begins by deducing and considering “faults” (use-related hazards) associated with device use (a “top-down” approach), whereas FMEA begins with the user interactions (a “bottom up” approach) and explores how they might lead to failure modes. As with FMEA, FTA is best accomplished by a diverse team using the brainstorming method. Even more than for FMEA, a task analysis is essential for constructing a FTA fault tree that includes all aspects of user–device interaction. Although FMEA and FTA are often used to identify and categorize use-related hazards, their effectiveness depends on the extent to which all hazards and use errors that could cause harm during device use can be deduced analytically by team members.
FTA, FMEA, and related approaches can be employed to identify and categorize use-related hazards, but the results should then be used to inform plans for simulated-use testing, which can confirm and augment the findings of the analytical risk analysis processes. Analytical processes do not include actual users or represent realistic use, and because use error is often “surprising” to analysts, simulated-use testing is necessary and should be designed to identify use errors not previously recognized or identified.
6.2 Identification of Known Use-Related Problems 🔗
When developing a new device, it is useful to identify use-related problems (if any) that have occurred with devices that are similar to the one under development with regard to use, the user interface or user interactions. When these types of problems are found, they should be considered during the design of the new device’s user interface. These devices might have been made by the same manufacturer or by other manufacturers. Sources of information on use-related problems include customer complaint files, and the knowledge of training and sales staff familiar with use-related problems. Information can also be obtained from previous HFE/UE studies conducted, for example, on earlier versions of the device being developed or on similar existing devices. Other sources of information on known use-related hazards are current device users, journal articles, proceedings of professional meetings, newsletters, and relevant internet sites, such as:
- FDA’s Manufacturer and User Facility Device Experience (MAUDE) database;
- FDA’s MedSun: Medical Product Safety Network;
- CDRH Medical Device Recalls;
- FDA Safety Communications;
- ECRI’s Medical Device Safety Reports;
- The Institute of Safe Medical Practices (ISMP's) Medication Safety Alert Newsletters; and
- The Joint Commission’s Sentinel Events.
All known use errors and use-related problems should be considered in the risk analysis for a new device and included if they apply to the new device.
6.3 Analytical Approaches to Identifying Critical Tasks 🔗
Analytical approaches involve review and assessment of user interactions with devices. These approaches are most helpful for design development when applied early in the process. The results include identification of hazardous situations, i.e. specific tasks or use scenarios including user-device interactions involving use errors that could cause harm. Analytical approaches can also be used for studying use-related hazardous situations that are too dangerous to study in simulated-use testing. The results are used to inform the formative evaluation (see Section 6.4.3) and human factors validation testing (see Section 8) that follow.
Analytical approaches for identifying use-related hazards and hazardous situations include analysis of the expected needs of users of the new device, analysis of available information about the use of similar devices, and employment of one or more analytical methods such as task analysis and heuristic and expert analyses. (Empirical approaches for identifying use-related hazards and hazardous situations include methods such as contextual inquiry and interview techniques and are discussed in Section 6.4.)
6.3.1 Task Analysis 🔗
Task analysis techniques systematically break down the device use process into discrete sequences of tasks. The tasks are then analyzed to identify the user interface components involved, the use errors that users could make and the potential results of all use errors. A simple example of a task analysis component for a hand-held blood glucose meter includes the tasks listed in Table 1.
Table 1. A simple task analysis for a hand-held blood glucose meter.
| # | Task |
|---|---|
| 1 | User places the test strip into the strip port of the meter |
| 2 | User lances a finger with a lancing device |
| 3 | User applies the blood sample to the tip of the test strip |
| 4 | The user waits for the meter to return a result |
| 5 | The user reads the displayed value |
| 6 | The user interprets the displayed value |
| 7 | The user decides what action to take next |
The task analysis can be used to help answer the following questions:
- What use errors might users make on each task?
- What circumstances might cause users to make use errors on each task?
- What harm might result from each use error?
- How might the occurrence of each use error be prevented or made less frequent?
- How might the severity of the potential harm associated with each use error be reduced?
Task analysis techniques can be used to study how users would likely perform each task and potential use error modes can be identified for each of the tasks. For each user interaction, the user actions can be identified using the model shown in Figure 4, i.e., the perceptual inputs, cognitive processing, and physical actions involved in performing the step. For example, perceptual information could be difficult or impossible to notice or detect and then as a cognitive component they could be difficult to interpret or could be misinterpreted; additional cognitive tasks could be confusing or complicated or inconsistent with the user’s past experiences; and physical actions could be incorrect, inappropriately timed, or impossible to accomplish. Each of these use error modes should be analyzed to identify the potential consequences of the errors and the potential resulting harm.
To begin to address the questions raised above, the analyst will need to understand more specific details such as:
- The effort required by the user to perform each task (e.g., to apply a blood sample to the test strip) correctly.
- The frequency that the user performs each task.
- The characteristics of the user population that might cause some users to have difficulty with each task.
- The characteristics of the use environment that might affect the test results or the user’s ability to perform each task.
- The impact of use errors on the accuracy, safety or effectiveness of the devices’ subsequent operations.
6.3.2 Heuristic Analysis 🔗
Heuristic analysis is a process in which analysts (usually HFE/UE specialists) evaluate a device’s user interface against user interface design principles, rules or “heuristic” guidelines. The object is to evaluate the user interface overall, and identify possible weaknesses in the design, especially when use error could lead to harm. Heuristic analyses include careful consideration of accepted concepts for design of the user interface. A variety of heuristics are available and you should take care to select the one or ones that are most appropriate for your specific application.
6.3.3 Expert Review 🔗
Expert reviews rely on clinical experts or human factors experts to analyze device use, identify problems, and make recommendations for addressing them. The difference between expert review and heuristic analysis is that expert review relies more heavily on assessment done by individuals with expertise in a specific area based on their personal experiences and opinions. The success of the expert review depends on the expert’s knowledge and understanding of the device technology, its use, clinical applications, and characteristics of the intended users, as well as the expert’s ability to predict actual device use. Reviews conducted by multiple experts, either independently or as a group, are likely to identify a higher number of potential use problems.
6.4 Empirical Approaches to Identifying Critical Tasks 🔗
Empirical approaches to identifying potential use-related hazards and hazardous situations derive data from users’ experiences interacting with the device or device prototypes or mock-ups. They provide additional information to inform the product development process beyond what is possible using analytical approaches.
Empirical approaches include methods such as contextual inquiry, interview techniques and simulated-use testing. To obtain valid data, it is important in such studies for the testing to include participants who are representative of the intended users. It is also important for facilitators to be impartial and to strive not to influence the behavior or responses of the participants. .
6.4.1 Contextual Inquiry 🔗
Contextual inquiry involves observing representatives of the intended users interacting with a currently marketed device (similar to the device being developed) as they normally would and in an actual use environment. The objective is to understand how design of the user interface affects the safety and effectiveness of its use, which aspects of the design are acceptable and which should be designed differently. In addition to observing, this process can include asking users questions while they use the device or interviewing them afterward. Users could be asked what they were doing and why they used the device the way they did. This process can help with understanding the users’ perspectives on difficult or potentially unsafe interactions, effects of the actual use environment, and various issues related to work load and typical work flow.
6.4.2 Interviews 🔗
Individual and group interviews (the latter are sometimes called “focus groups”) generate qualitative information regarding the perceptions, opinions, beliefs and attitudes of individual or groups of device users and patients. In the interviews, users can be asked to describe their experiences with existing devices, specific problems they had while using them, and provide their perspectives on the way a new device should be designed.
Interviews can focus on topics of particular interest and explore specific issues in depth. They should be structured to cover all relevant topics but allow for unscripted discussion when the interviewee’s responses require clarification or raise new questions. Individual interviews allow the interviewer to understand the perspectives of individuals who, for example, might represent specific categories of users or understand particular aspects of device use or applications. Individual interviews can also make it easier for people to discuss issues that they might not be comfortable discussing in a group. Group interviews offer the advantage of providing individuals with the opportunity to interact with other people as they discuss topics.
6.4.3 Formative Evaluations 🔗
Formative evaluations are used to inform device user interface design while it is in development. It should focus on the issues that the preliminary analyses indicated were most likely to involve use safety (e.g., aspects of user interaction with the device that are complicated and need to be explored). It should also focus on those areas where design options for the user interface are not yet final.
Formative evaluation complements and refines the analytical approaches described in Section 6.3, revealing use issues that can only be identified through observing user interaction with the device. For example, formative evaluation can reveal previously unrecognized use-related hazards and use errors and help identify new critical tasks. It can also be used to:
- Inform the design of the device user interface (including possible design tradeoffs),
- Assess the effectiveness of measures implemented to reduce or eliminate use-related hazards or potential use errors,
- Determine training requirements and inform the design of the labeling and training materials (which should be finalized prior to human factors validation testing), and
- Inform the content and structure of the human factors validation testing.
The methods used for formative evaluation should be chosen based on the need for additional understanding and clarification of user interactions with the device user interface. Formative evaluation can be conducted with varying degrees of formality and sample sizes, depending on how much information is needed to inform device design, the complexity of the device and its use, the variability of the user population, or specific conditions of use (e.g., worst-case conditions). Formative evaluations can involve simple mock-up devices, preliminary prototypes or more advanced prototypes as the design evolves. They can also be tailored to focus on specific accessories or elements of the user interface or on certain aspects of the use environment or specific sub-groups of users.
Design modifications should be implemented and then evaluated for adequacy during this phase of device development in an iterative fashion until the device is ready for human factors validation testing. User interface design flaws identified during formative evaluation can be addressed more easily and less expensively than they could be later in the design process, especially following discovery of design flaws during human factors validation testing. If no formative evaluation is conducted and design flaws are found in the human factors validation testing, then that test essentially becomes a formative evaluation.
The effectiveness of formative evaluation for providing better understanding of use issues (and preventing a human factors validation test from becoming a formative evaluation) will depend on the quality of the formative evaluation. Depending on the rigor of the test you conduct, you might underestimate the existence or importance of problems found, for example, because the test participants were unrealistically well trained, capable, or careful during the test. Unlike human factors validation testing, company employees can serve as participants in formative evaluation; however, their performance and opinions could be misleading or incomplete if they are not representative of the intended users, are familiar with the device or are hesitant to express their honest opinions.
The protocol for a formative evaluation typically specifies the following:
- Evaluation purpose, goals and priorities;
- Portion of the user interface to be assessed;
- Use scenarios and tasks involved;
- Evaluation participants;
- Data collection method or methods (e.g., cognitive walk-through, observation, discussion, interview);
- Data analysis methods; and
- How the evaluation results will be used.
The results of formative evaluation should be used to determine whether design modifications are needed and what form they should take. Because this testing is conducted on a design in progress, is often less formal and often uses different methods, the results will not apply directly to the final user interface design.
Formative evaluations can be effective tools for identifying and understanding ways in which the user interface affects user interactions. The quality of the test results and the information gained from them will depend on the quality of the formative evaluation. You should take care not underestimate or overestimate the frequency of problems based on the formative evaluation results. Participants could be unrealistically well trained, capable, or careful during the test or the device prototype could differ from the final design in ways that affect user interactions.
6.4.3.1 Cognitive Walk-Through 🔗
A simple kind of formative evaluation involving users is the cognitive walk-through. In a cognitive walk-through, test participants are guided through the process of using a device. During the walk-through, participants are questioned and encouraged to discuss their thought processes (sometimes called “think aloud”) and explain any difficulties or concerns they have.
6.4.3.2 Simulated-Use Testing 🔗
Simulated-use testing provides a powerful method to study users interacting with the device user interface and performing actual tasks. This kind of testing involves systematic collection of data from test participants using a device, device component or system in realistic use scenarios but under simulated conditions of use (e.g., with the device not powered or used on a manikin rather than an actual patient). In contrast to a cognitive walk-through, simulated-use testing allows participants to use the device more independently and naturally. Simulated use testing can explore user interaction with the device overall or it can investigate specific human factors considerations identified in the preliminary analyses, such as infrequent or particularly difficult tasks or use scenarios, challenging conditions of use, use by specific user populations, or adequacy of the proposed training.
During formative evaluation, the simulated-use testing methods can be tailored to suit your needs for collecting preliminary data. Data can be obtained by observing participants interacting with the device and interviewing them. Automated data capture can also be used if interactions of interest are subtle, complex, or occur rapidly, making them difficult to observe. The participants can be asked questions or encouraged to “think aloud” while they use the device. They should be interviewed after using the device to obtain their perspectives on device use, particularly related to any use problems that occurred, such as obvious use error. The observation data collection can also include any instances of observed hesitation or apparent confusion, can pause to discuss problems when they arise, or include other data collection methods that might be helpful to inform the design of a specific device user interface.
7. Elimination or Reduction of Use-Related Hazards 🔗
Use-related device hazards should be identified through preliminary analyses and evaluations (Section 6). When identified, these hazards should be, to the extent possible, controlled through elimination of the hazard (designed out), reduction in likelihood or reduction in the severity of the resulting harm prior to initiating the human factors validation test. Use-related hazards are addressed by applying risk management strategies. Often, any given strategy may be only partially effective and multiple strategies may be necessary to address each use-related hazard. ANSI/AAMI/ISO 14971 lists the following risk management options in order of preference and effectiveness:
1. Inherent safety by design – For example:
- Use specific connectors that cannot be connected to the wrong component.
- Remove features that can be mistakenly selected or eliminate an interaction when it could lead to use error.
- Improve the detectability or readability of controls, labels, and displays.
- Automate device functions that are prone to use error when users perform the task manually.
2. Protective measures in the medical device itself or in the manufacturing process – For example:
- Incorporate safety mechanisms such as physical safety guards, shielded elements, or software or hardware interlocks.
- Include warning screens to advise the user of essential conditions that should exist prior to proceeding with device use, such as specific data entry.
- Use alerts for hazardous conditions, such as a “low battery” alert when an unexpected loss of the device’s operation could cause harm or death.
- Use device technologies that require less maintenance or are “maintenance free.”
3. Information for safety – For example:
- Provide written information, such as warning or caution statements in the user manual that highlight and clearly discuss the use-related hazard.
- Train users to avoid the use error.
Design modifications to the device and its user interface are generally the most effective means for eliminating or reducing use-related hazards. If design modifications are not possible or not practical, it might be possible to implement protective measures, such as reducing the risk of running out of battery power by adding a “low battery” alert to the device or using batteries with a longer charge life. Device labeling (including the instructions for use) and training, if designed adequately, can support users to use devices more safely and effectively and are important HFE/UE strategies to address device use hazards. These strategies are not the most preferred, though, because they rely on the user to remember or refer back to the information, labeling might be unavailable during use, and knowledge gained through training can decay over time. Nonetheless, unless a device design modification can completely remove the possibility of a use error, the labeling and training (if applicable) should also be modified to address the hazard: if no other options are available, users should at least be given sufficient information to understand and avoid the hazard.
Regardless of the risk management strategies used, they should be tested to ensure that use-related hazards were successfully addressed and new hazards were not introduced.
8. Human Factors Validation Testing 🔗
Human factors validation testing4 is conducted to demonstrate that the device can be used by the intended users without serious use errors or problems, for the intended uses and under the expected use conditions. The testing should be comprehensive in scope, adequately sensitive to capture use errors caused by the design of the user interface, and should be performed such that the results can be generalized to actual use. https://www.notion.so/innolitics/4-801f24e7710f4249aa0e4e1dd6c18af5?source=copy_link
The human factors validation testing should be designed as follows:
- The test participants represent the intended (actual) users of the device.
- All critical tasks are performed during the test.
- The device user interface represents the final design.
- The test conditions are sufficiently realistic to represent actual conditions of use.
For the device to be considered to be optimized with respect to use safety and effectiveness, the human factors validation testing should be sufficiently sensitive to capture use-related problems resulting from user interface design inadequacies, whether or not the users are aware of having made use errors. Furthermore, the human factors validation test results should show no use errors or problems that could result in serious harm and that could be eliminated or reduced through modification of the design of the user interface, using one or more of the measures listed in Section 7.
The realism and completeness of the human factors validation testing should support generalization of the results to demonstrate the device’s use safety and effectiveness in actual use. The test protocol should include discussion of the critical tasks (identified based on the potential for serious harm caused by use error; see Section 6.1) and the methods used to collect data on the test participants’ performance and subjective assessment of all critical tasks. The results of the testing should facilitate analysis of the root causes of use errors or problems found during the testing.
Human factors validation testing is generally conducted under conditions of simulated use, but when necessary, human factors data can also be collected under conditions of actual use or as part of a clinical study (see Section 8.3). You should perform human factors validation testing under conditions of actual use when simulated-use test methods are inadequate to evaluate users’ interactions with the device. This determination should be based on the results of your preliminary analyses (see Section 6).
FDA encourages manufacturers to submit a draft of the human factors testing protocol prior to conducting the test so we can ensure that the methods you plan to use will be acceptable. The premarket mechanism for this is a Pre-submission (see Requests for Feedback on Medical Device Submissions: The Pre-Submission Program and Meetings with FDA Staff).
8.1 Simulated-Use Human Factors Validation Testing 🔗
The conditions under which simulated-use testing is conducted should be sufficiently realistic so that the results of the testing are generalizable to actual use. The need for realism is therefore driven by the analysis of risks related to the device’s specific intended use, users, use environments, and the device user interface. To the extent that environmental factors might affect users’ interactions with elements of the device user interface, they should be incorporated into the simulated use environment (e.g., dim lighting, multiple alarm conditions, distractions, and multi-tasking).
During simulated-use human factors validation testing, test participants should be given an opportunity to use the device as independently and naturally as possible, without interference or influence from the test facilitator or moderator. Use of the “think aloud” technique (in which test participants are asked to vocalize what they are thinking while they use the device), although perhaps useful in formative evaluation, is not acceptable in human factors validation testing because it does not reflect actual use behavior. If users would have access to the labeling in actual use, it should be available in the test; however, the participants should be allowed to use it as they choose and should not be instructed to use it. Participants may be asked to evaluate the labeling as part of the test, but this evaluation should be done separately, after the simulated-use testing is completed. If the users would have access to a telephone help line, it may be provided in the test but it should be as realistic as possible; e.g., the telephone assistant should not be in the room and should not guide the users through the specific test tasks.
8.1.1 Test Participants (Subjects) 🔗
The most important consideration for test participants in human factors validation testing is that they represent the population of intended users.
The number of test participants involved in human factors validation depends on the purpose of the test. For human factors validation, sample size is best determined from the results of the preliminary analyses and evaluations. Manufacturers should make their own determinations of the necessary number of test participants but, in general, the minimum number of participants should be 15. Note that the recommended minimum number of participants could be higher for specific device types. (See Appendix B for a discussion of sample size considerations.)
If the device has more than one distinct population of users, then the validation testing should include at least 15 participants from each user population. The FDA views user populations as distinct when their characteristics would likely affect their interactions with the device or when the tasks they perform on the device would be different. For example, some devices will have users in different age categories (pediatric, adolescent, adult, or geriatric) or users in different professional categories (e.g., health care provider, lay user); other devices will have users with different roles (e.g., installers, healthcare providers with unique specialties, or maintenance personnel).
The human factors validation test participants should be representative of the range of characteristics within their user group. The homogeneity or heterogeneity of user groups can be difficult to establish precisely but you should include test participants that reflect the actual user population to the extent possible. If intended users include a pediatric population, the testing should include a group of representative pediatric users; when a device is intended to be used by both pediatric and adult users, FDA views these as distinct populations. Likewise, if a device is intended to be used by both professional healthcare providers and lay users, FDA views these as distinct user populations. In many cases, the identification of distinct user groups should be determined through the preliminary analyses and evaluations (Section 6). For instance, if different user groups will perform different tasks or will have different knowledge, experience or expertise that could affect their interactions with elements of the user interface and therefore have different potential for use error, then these users should be separated into distinct user populations (each represented by at least 15 test participants) for the purpose of validation testing. The ways in which users differ from one another are unlimited, so you should focus on user characteristics that could have a particular influence on their interactions with elements of the device user interface, such as age, education or literacy level, sensory or physical impairments or occupational specialty.
If the device is intended to treat patients who have a medical condition that can cause them to have functional limitations, people with a representative range of those limitations should be considered during preliminary evaluations and included as representative users in the human factors validation testing. For example, people who use diabetes management devices might have retinopathy or neuropathy caused by diabetes. If you choose not to design your device to accommodate the needs of people with functional limitations who would otherwise be likely to use your device, your labeling should clearly explain the capabilities users need to have to use the device safely and effectively.
Note, to minimize potential bias introduced into your validation testing, your employees should not serve as test participants in human factors validation testing except in rare cases when all users necessarily are employees of the manufacturer (e.g., specialized service personnel).
For the results of the human factors validation testing to demonstrate safe and effective use by users in the United States, the participants in the testing should reside in the US. Studies performed in other countries or with non-US residents may be affected (positively or negatively) by different clinical practices that exist in other countries, different units of measure used, language differences that change the way labeling and training are understood, etc. Exceptions to this policy will be considered on a case-by-case basis and will be based on a sound rationale that considers the relevant differences from conditions in the US. In addition to the user interface of the device, the labeling and training should correspond exactly to that which would be used for the device if marketed in the US.
8.1.2 Tasks and Use Scenarios 🔗
The human factors validation testing should include all critical tasks identified in the preliminary analyses and evaluations. Tasks that logically occur in sequence when using the device (e.g., when performing device set-up, data entry or calibration) can be grouped into use scenarios, which should be described in the test protocol. Use scenarios in the testing should include all necessary tasks and should be organized in a logical order to represent a natural workflow. Devices associated with a very large number of critical tasks might need to be assessed in more than one human factors validation test session (e.g., with the same participants or different participants who are representative of the same user population). Prior to testing, you should define user performance that represents success for each task.
The test protocol should also provide a rationale for the extent of device use and the number of times that participants will use the device. For example, for devices like over-the-counter automatic external defibrillators (AEDs), only one use session should be conducted since additional attempts would be irrelevant in an actual use setting. For devices that are used frequently and have a learning curve that requires repeated use to establish reasonable proficiency, allowing the participant to use the device multiple times during a test session might be appropriate (but performance and interview data should be collected for each use). For other devices, typical use might involve repeated performance of critical tasks and so multiple performances of those tasks should be included in the test protocol.
Critical tasks or use scenarios involving critical tasks that have a low frequency of occurrence require careful consideration and those tasks should be included in the testing as appropriate to the risk severity. Rare or unusual use scenarios for which use errors could cause serious harm are an important consideration for testing safe and effective medical device use. Infrequent but hazardous use scenarios can be difficult to identify, which underscores the necessity for careful application of the preliminary analyses and evaluations.
8.1.3 Instructions for Use 🔗
The design of the device labeling can be studied in formative evaluation, but the labeling used in the human factors validation testing should represent the final designs. This applies to the labels on the device and any device accessories, information presented on the device display, the device packaging and package labels, instructions for use, user manuals, package inserts, and quick-start guides.
The human factors validation testing can indirectly serve to assess the adequacy of the instructions for use for the device, but only in the context of use of the device, including the participants’ understanding or “knowledge” regarding critical issues of use. The goal is to determine the extent to which the instructions for use support the users’ safe and effective use of the device. If the device labeling is inadequate, it will be evidenced by participant performance or subjective feedback. If the results of human factors validation testing include use errors on critical tasks or participant feedback indicating difficulty with critical tasks, stating in a premarket submission that you mitigated the risks by modifying the instructions for use or some other element of labeling is not acceptable unless you provide additional test data demonstrating that the modified elements were effective in reducing the risks to acceptable levels.
8.1.4 Participant Training 🔗
The design and extent of the training that needs to be provided to users can also be studied in formative evaluation, but the training provided to the human factors validation test participants should approximate the training that actual users would receive. If you anticipate that most or all users would receive minimal or no training, then the test participants in the human factors validation test should not be trained. If the results of human factors validation testing include use errors on critical tasks or subjective responses indicating difficulty with critical tasks, stating in a premarket submission your intention to mitigate the risks by providing “additional training” is not acceptable unless you provide additional data that demonstrates that it would be effective in reducing the risks to acceptable levels.
To the extent practicable, the content, format, and method of delivery of training given to test participants should be comparable to the training that actual users would receive. A human factors validation test conducted after participants have been trained differently than they would be in actual use is not valid. Because retention of training decays over time, testing should not occur immediately following training; some period of time should elapse. In some cases, giving the participants a break of an hour (e.g., a “lunch break”) is acceptable; in other cases, a gap of one or more days would be appropriate, particularly if it is necessary to evaluate training decay as a source of use-related risk. For some types of devices used in non-clinical environments (e.g., the home), it might be reasonable to allow the participants to take the instructions for use home with them after the training session to review as they choose before the test session. The test protocol should describe the training provided for the testing, including the content and delivery modes and the length of time that elapsed prior to testing.
8.1.5 Data Collection 🔗
The human factors validation test protocol should specify the types of data that will be collected in the test. Some data is best collected through observation; for example, successful completion of or outcome from critical tasks should be measured by observation rather than relying solely on participant opinions. Although measuring the time it takes participants to conduct a specific task might be helpful for purposes such as comparing the ease of use of different device models, performance time is only considered to be a meaningful measure of successful performance of critical tasks if the speed of device use is clinically relevant (e.g., use of an automated external defibrillator). Timing of tasks that have not been defined in advance as being time-critical should not be included in human factors validation testing. Some important aspects of use cannot be assessed through task performance and instead require direct questioning of the participant to ascertain their understanding of essential information. Observational and knowledge task data should be supplemented with subjective data collected through interviews with the participants after the use scenarios are completed.
8.1.5.1 Observational Data 🔗
The human factors validation testing should include observations of participants’ performance of all the critical use scenarios (which include all the critical tasks). The test protocol should describe in advance how test participant use errors and other meaningful use problems were defined, identified, recorded and reported. The protocol should also be designed such that previously unanticipated use errors will be observed and recorded and included in the follow-up interviews with the participants.
Observational data recorded during the testing should include use problems, and most importantly use errors, such as a test participant failing the task of priming an intravenous line without disconnecting the line from the simulated “patient” or not finding a vital control on the user interface when it is required for successful task performance.
“Close calls” are instances in which a user has difficulty or makes a use error that could result in harm, but the user takes an action to “recover” and prevents the harm from occurring. Close calls should be recorded when they are observed and discussed with the test participants after they have completed all the use scenarios. In addition, repeated attempts to complete a task and apparent confusion could indicate potential use error and therefore should also be collected as observational data and discussed during the interviews with test participants.
8.1.5.2 Knowledge Task Data 🔗
Many critical tasks are readily evaluated through simulated-use techniques and use errors are directly observable, enabling user performance to be assessed through observation during simulated use testing. However, other critical tasks cannot be evaluated this way because they involve users’ understanding of information, which is difficult to ascertain by observing user behavior. For instance, users might need to understand critical contraindication and warning information. Lay users might need to understand a device’s vulnerabilities to specific environmental hazards, the potential harm resulting from taking shortcuts or reusing disposable components, or the need to periodically perform maintenance on the device or its accessories. It might be vital for a healthcare provider to know that a device should never be used in an oxygen-rich environment but testing under conditions of simulated use would be difficult since establishing that the test environment was oxygen-rich during the testing and then asking users to use the device and observing their behavior would likely not produce meaningful results.
The user interface components involved in knowledge tasks are usually the user manual, quick start guide, labeling on the device itself, and training. The user perceives and processes the information provided and if these components are well designed, this information becomes part of the user’s “knowledge.” This knowledge can be tested by questioning the test participants. The questions should be open-ended and worded neutrally.
8.1.5.3 Interview Data 🔗
The observation of participant performance of the test tasks and assessment of their understanding of essential information (if applicable) should be followed by a debriefing interview. Interviews enable the test facilitator to collect the users’ perspectives, which can complement task performance observations but cannot be used in lieu of them. The two data collection methods generate different types of information, which might reinforce each other, such as when the interview data confirm the test facilitator’s observations. At other times, the two sources of information might conflict, such as when the participant’s reported reasons for observed actions are different from the reasons presumed by the observer. For instance, the user might have made several use errors but when interviewed might have no complaints and might not have noticed making any errors. More often, the user might make no use errors on critical tasks but in the interview might point out one or more aspects of the user interface that were confusing or difficult and that could have caused problems.
In the interview, the participant should provide a subjective assessment of any use difficulties experienced during the test (e.g., confusing interactions, awkward manual manipulations, unexpected device operation or response, difficulty reading the display, difficulty hearing an alarm, or misinterpreting, not noticing or not understanding a device label). The interview should be composed of open-ended and neutrally-worded questions that start by considering the device overall and then focus on each critical task or use scenario. You should investigate all use errors in the post-test debriefing interview with the participant to determine how and why they believe the error occurred. For example:
- “What did you think of the device overall?”
- “Did you have any trouble using it? What kind of trouble did you have?”
- “Was anything confusing? What was confusing?”
- “Please tell me about this [use error or problem observed]. What happened? How did that happen?”
- Note: The interview should include this question for each use error or problem observed for that test participant.
It is important for the interviewer to accept all participant responses and comments without judgment so as to obtain the participants’ true perspectives and not to influence their responses.
8.1.6 Analysis of Human Factors Validation Test Results 🔗
The results of the human factors validation testing should be analyzed qualitatively to determine if the design of the device (or the labeling or user training) needs to be modified to reduce the use-related risks to acceptable levels. To do this, the observational data and knowledge task data should be aggregated with the interview data and analyzed carefully to determine the root cause of any use errors or problems (e.g., “close calls” and use difficulties) that occurred during the test. The root causes of all use errors and problems should then be considered in relation to the associated risks to ascertain the potential for resulting harm and determine the priority for implementing additional risk management measures. Appendix C of this document presents sample analyses of human factors validation test results.
Depending on the extent of the risk management strategies implemented, retesting might be necessary. You should address aspects of the user interface that led to use errors and problems with critical tasks by designing and implementing risk management strategies. You might find it useful to conduct additional preliminary analyses and evaluations (Section 6) to explore and finalize the modifications. You should then conduct human factors validation testing on the modified use interface elements to assess the success of the risk management measures at reducing risks to acceptable levels without introducing any new unacceptable risks. If the modified elements affect only some aspects of device use, the testing can focus on those aspects of use only.
8.1.7 Residual Risk 🔗
It is practically impossible to make any device error-proof or risk-free; some residual risk will remain, even if best practices were followed in the design of the user interface. All risks that remain after human factors validation testing should be thoroughly analyzed to determine whether they can be reduced or eliminated. True residual risk is beyond practicable means of elimination or reduction through modifications to the user interface, labeling, or training. Human factors validation testing results indicating that serious use errors persist are not acceptable in premarket submissions unless the results are analyzed well and the submission shows that further reduction of the errors’ likelihood is not possible or practical and that the benefits of device use outweigh the residual risks.
The analysis of use-related risk should determine how the use errors or problems occurred within the context of device use, including the specific aspect of the user interface that caused problems for the user. This analysis should determine whether design modifications are needed, would be possible and might be effective at reducing the associated risks to acceptable levels. Indeed, test participants often suggest design modifications when they are interviewed within a human factors validation test. Use errors or problems associated with high levels of residual risk should be described in the human factors validation report. This description should include how the use problems were related to the design of the device user interface. If your analyses show that design modifications are needed but would be impossible or impractical to implement, you should explain this and describe how the overall benefits of using the device outweigh the residual risks.
If design flaws that could cause use errors that could result in harm are identified and could be reduced or eliminated through design changes, stating in a premarket submission that you plan to address them in subsequent versions of the device is not acceptable. Note also that finding serious use errors and problems during human factors validation testing might indicate that insufficient analysis, formative evaluation, and modification of the device user interface was undertaken during design development.
8.2 Human Factors Validation Testing of Modified Devices 🔗
When a manufacturer has modified a device already on the market, the risk analysis should include all aspects of the device that were modified and all elements of the device that were affected by the modifications. The risk analysis should also include all aspects of the users’ interactions with the device that were affected by the modifications, either directly or indirectly.
As with any other device, the need to conduct an additional human factors validation test should be based on the risk analysis of the modifications made and if the use-related risk levels are unacceptable, the test should focus on those hazard-related use scenarios and critical tasks. The test may, however, be limited to assessment of those aspects of users’ interactions and tasks that were affected by the design modifications.
When a manufacturer is modifying a currently marketed device in response to use-related problems, possibly as part of a Corrective and Preventive Action (CAPA) or recall, the human factors validation test should evaluate the modified user interface design using the same methods as usual. However, the evaluation will be most effective if it also involves direct solicitation of the user’s comparison of the design modification to the previous design. The test administrator should explain the known problems and then show the participant the previous version of the interface component along with the new or modified version. The participants should then be asked questions, such as:
- “Do you believe the new design is better than the old one? Please tell me how the new one is [better/worse] than the old one.”
- “How effective do you think these modifications will be in preventing the use error from occurring? Please tell me why you think it [will/will not].”
- “Could these changes cause any other kind of use difficulty? What kinds of difficulty?”
- “Are these modifications sufficient or does this need further modification? How should it be modified?”
8.3 Actual Use Testing 🔗
Due to the nature of some types of device use and use environments that can be particularly complicated or poorly understood, it might be necessary to test a device under conditions of actual use. For example, it would be impossible to test some aspects of a prosthetic limb or a hearing aid programming device under simulated use conditions; and the results of testing a home dialysis machine in a conference room might not be generalizable to use of the device in a residential environment.
Human factors testing performed under actual use conditions should be preceded by appropriate simulated-use testing to ensure that the device is sufficiently well designed to be safe in actual use (to the degree that simulated-use testing can provide such assurance).
Actual-use human factors testing should follow the same general guidelines as simulated use human factors validation testing, described in Section 8.1; noting that when actual use testing is needed to determine safety and effectiveness of the proposed device and the requirements outlined in 21 CFR §812 apply, then an Investigational Device Exemption (IDE) is needed.5 In such a test, the test participants should be representative of the actual users, the clinical environments should be representative of the actual use environments and the testing process should affect the participants’ interactions with the device as little as possible.
Actual-use testing can also be conducted as part of a clinical study. However, in a clinical study, the participants are generally trained differently and are more closely supervised than users would be in real-world use, so the resulting data (e.g., observations and interviews) should be viewed in that context. Another way in which a clinical trial differs from a simulated-use human factors validation test is that the sample sizes are generally much larger in order for the outcome data to be statistically significant. For studies in which the test participants use the device at home, opportunities for direct observation can be limited; regardless, it is inadequate to depend solely on self-reports of device use to understand the users’ interactions with the device because these data can be incomplete or inaccurate. To the extent practicable, such data should be supplemented with observational data.
You should consult with your internal institutional review board for the protection of human subjects (IRB) to determine the need to implement specific safeguards of test participant safety and personal privacy, including informed consent forms.
For more information about Investigational Device Exemptions, see FDA’s guidance, FDA Decisions for Device Exemption (IDE) Clinical Investigations. For more information about pivotal clinical studies, see FDA’s guidance, Design Considerations for Pivotal Clinical Investigations for Medical Devices.
9. Documentation 🔗
Documenting your risk management, HFE/UE testing, and design optimization processes (e.g., in your design history file as part of your design controls) provides evidence that you considered the needs of the intended users in the design of your new device and determined that the device is safe and effective for the intended users, uses and use environments. When it is required, providing information about these processes as part of a premarket submission for a new device will reduce the need for requests for additional information and facilitate FDA’s review of all HFE/UE information contained in your submission. A sample outline of a HFE/UE report that could be submitted to FDA is shown in Appendix A. The report should provide a summary of the evaluations performed and enough detail to enable the FDA reviewer to understand your methods and results, but the submission would not need to include, for example, all the raw data from a human factors validation test. All documentation related to HFE/UE processes, whether required to be submitted to FDA or not, should be kept in manufacturers’ files.
10. Conclusion 🔗
The advantages of optimizing device design through application of HFE/UE extend beyond improved safety. Many device manufacturers have found that the application of HFE/UE during the development of their products reduces the need for design modifications and costly updates after market introduction and offers competitive advantages. With increased safety, the likelihood of your incurring expenses associated with product recalls or liability is reduced; and when HFE/UE approaches are used during the design development process, particularly if the perspective of users is taken into account, the overall ease of use and appeal of a device can simultaneously be enhanced.
Appendix A HFE/UE Report 🔗
A HFE/UE report included in a premarket submission should provide information pertaining to device use safety and effectiveness in summary form. The report should discuss the safety-related HFE/UE considerations, issues, processes, resolutions, and conclusions. The level of detail of documentation submitted should be sufficient to describe your identification, evaluation, and final assessment of all serious use-related hazards for the device. To facilitate FDA review, materials used directly in the HF/UE process, including portions of risk analyses focusing on user interactions with the device and specific risk analysis processes, results and conclusions should be included in the HFE/UE report. If necessary, the report may refer to materials relevant to the HFE/UE process located in other parts of a submission.
A recommended structure for the HFE/UE report, which will support efficient FDA review of these materials, is listed in Table A-1 and described in the text that follows.
Table A-1. Outline of HFE/UE Report
Sec. Contents
-
Conclusion The <device> has been found to be safe and effective for the intended users, uses and use environments.
- Brief summary of HFE/UE processes and results that support this conclusion
- Discussion of residual use-related risk
-
Descriptions of intended device users, uses, use environments, and training
- Intended user population(s) and meaningful differences in capabilities between multiple user populations that could affect user interactions with the device
- Intended use and operational contexts of use
- Use environments and conditions that could affect user interactions with the device
- Training intended for users
-
Description of device user interface
- Graphical representation of device and its user interface
- Description of device user interface
- Device labeling
- Overview of operational sequence of device and expected user interactions with user interface
-
Summary of known use problems
- Known use problems with previous models of the subject device
- Known use problems with similar devices, predicate devices or devices with similar user interface elements
- Design modifications implemented in response to post-market use error problems
-
Analysis of hazards and risks associated with use of the device
- Potential use errors
- Potential harm and severity of harm that could result from each use error
- Risk management measures implemented to eliminate or reduce the risk
- Evidence of effectiveness of each risk management measure
-
Summary of preliminary analyses and evaluations
- Evaluation methods used
- Key results and design modifications implemented in response
- Key findings that informed the human factors validation test protocol
-
Description and categorization of critical tasks
- Process used to identify critical tasks
- List and descriptions of critical tasks
- Categorization of critical tasks by severity of potential harm
- Descriptions of use scenarios that include critical tasks
-
Details of human factors validation testing
- Rationale for test type selected (i.e., simulated use, actual use or clinical study)
- Test environment and conditions of use
- Number and type of test participants
- Training provided to test participants and how it corresponded to real-world training levels
- Critical tasks and use scenarios included in testing
- Definition of successful performance of each test task
- Description of data to be collected and methods for documenting observations and interview responses
- Test results: Observations of task performance and occurrences of use errors, close calls, and use problems
- Test results: Feedback from interviews with test participants regarding device use, critical tasks, use errors, and problems (as applicable)
- Description and analysis of all use errors and difficulties that could cause harm, root causes of the problems, and implications for additional risk elimination or reduction
Section 1: Conclusion
The report should begin with a conclusion stating that the new medical device has been found to be safe and effective for the intended users, uses, and use environments. The conclusion should be supported by a summary of the HFE/UE processes conducted (e.g., HFE/UE analyses and evaluations, design modifications, and validation testing) and analysis of the results. This section should discuss any residual use-related risk that remained after the human factors validation testing. If applicable, this section should provide a sound rationale that modifications to the user interface (including the device and the labeling) would not further reduce risk, are not possible or not practicable, and the remaining residual userelated risks are outweighed by the benefits derived from use of the device.
Section 2: Descriptions of intended device users, uses, use environments, and training
This section should include:
- A description of the intended user population or, if there is more than one distinct user population, each population; the description should include meaningful differences in capabilities or use responsibilities between user populations that could affect their interactions with the device (such as lay and professional users who might use the same device to perform different tasks or different types of professionals who might perform different tasks on the device);
- A summary of the device’s intended use;
- A summary of the device’s operational context of use (such as the requirement that a user be trained by a nurse prior to using the device, or it is used in an operating room, or it is used differently for different applications) and critical aspects of device operation, such as set up, maintenance, cleaning, reprocessing;
- A summary of the intended use environments (e.g., hospital, medevac vehicle, non-clinical environment) and the characteristics of those environments (e.g., glare, vibration, ambient noise, high levels of activity) that could affect user interactions with the device; and
- A description of any training users would receive; a sample of the training materials (such as a DVD, computer slides, or a pamphlet) may be appended to the report.
Section 3: Description of device user interface
This section should include (as applicable):
- A graphical representation (e.g., photographs, illustrations or line drawings) of the device and its user interface, including a depiction of the overall device and all components of the user interface with which the user will interact (e.g., display and function screens, alarm speakers, controls, keypads, dedicated buttons, doors, components to be connected, retaining clips);
- A written description of the device user interface;
- A copy of the labeling materials that will be provided to the user with the device (e.g., instructions for use, user manual, quick-start guides, packaging); and
- An overview of the operational sequence of the device and the users’ expected interactions with the user interface, consisting of the sequence of user actions performed to use the device (and resulting device responses, as appropriate).
Section 4: Summary of known use problems
This section should include all use problems known to exist in previous models of the same device (as applicable) or with similar types of medical devices (e.g., predicate devices). In some cases, no use problems are known to exist and if so, this may be stated. If the submission is for a device that has been modified specifically in response to use problems that occurred in the field, this section should highlight those problems and the new modifications.
Section 5: Analysis of hazards and risks associated with use of the device
This section should provide an excerpt from the comprehensive risk analysis that contains all the use-related hazards and risks, including those associated with potential use errors. The section should include at least one hazardous scenario associated with each use error, the potential harm that could result, the potential severity of the harm, all risk control measures implemented to eliminate or reduce the risk, and the source of evidence that each risk control measure was effective.
Section 6: Summary of preliminary analyses and evaluations
This section should identify the preliminary analysis and evaluation methods used (e.g., specific analysis techniques, formative evaluations), summarize the key results of those analyses and evaluations, describe any modifications that were implemented to the user interface design in response to the results, and discuss the key findings that informed development of the protocol for the human factors validation test.
Section 7: Description and categorization of critical tasks
This section should explain the process that was followed to identify the critical tasks during the preliminary analyses and evaluations; it should also provide a list and descriptions of the critical tasks. The section should identify the severity of the potential harm that could result from use errors on the critical tasks. The section should also describe the use scenarios to be included in the human factors validation test and list the critical tasks and other tasks that constitute each use scenario.
Section 8: Details of the human factors validation testing
This section should include a synopsis of all activities conducted. The section should contain a summary of the test results, a comprehensive analysis of all use errors and problems that occurred that could have resulted in harm in real-world use, a description of all design modifications made to the user interface in response to the test results, and a risk-benefit discussion. A full test protocol and a sample of all scripts and forms used in the testing should be appended to the report.
Appendix B Considerations for Determining Sample Sizes for Human Factors Validation Testing 🔗
Published estimates of the number of test participants required to identify all problems that exist in a user interface6 are based on a set of assumptions regarding: a fixed (and known) probability of encountering a problem, a uniform likelihood for each participant to encounter each problem, and the independence of the problems (that is, encountering one problem will not increase or decrease the likelihood of finding other problems). However, none of these assumptions reflects the real world. Most importantly, individual likelihoods of encountering a problem with a user interface vary considerably, depending on the user’s personal capabilities, knowledge and experience levels, nature of interaction with the device, frequency of task performance, attributes of the use environment and use conditions, and the nature of the problem. Theoretically, the lower the chances of finding a problem (e.g., if the problem is subtle or the users are highly skilled), the more people you need to test to provide reasonable assurance that the problem will be identified. In practice, it is difficult to identify all the problems in a new user interface and this is, in fact, one of the reasons for conducting human factors validation testing. Even for those problems that are known or believed to exist, it is difficult to anticipate how likely it is that the problems will be detected or cause observable use errors or problems or to anticipate the variability among test participants populations prior to testing. Consequently, it would be extremely difficult to develop a formula for the statistically “correct” sample size for testing a specific device.
Faulkner (2003) conducted a study that collected empirical data from a sample of 60 individuals with varying levels of experience with computers in general and with the software used in the test specifically. The results suggested that a sample of 15 people was sufficient to find a minimum of 90% and an average of 97% of all problems with that software; a sample of 20 people was able to find a minimum of 95% and an average of 98% of the problems (see Table B-1 and Figure B-1). Note that the change in detection rate decreases asymptotically to zero as the number of users increases, and a sample of 30 users detected an average of less than 2% more problems than did a sample of 15 users.
Table B-1. Percentage of Total Known Usability Problems Found in 100 Analysis Samples (Faulkner, 2003).
| No. users | Min. % Found | Mean % Found | SD | SE |
|---|---|---|---|---|
| 5 | 55 | 85.55 | 9.2957 | .9295 |
| 10 | 82 | 94.69 | 3.2187 | .3218 |
| 15 | 90 | 97.05 | 2.1207 | .2121 |
| 20 | 95 | 98.4 | 1.6080 | .1608 |
| 30 | 97 | 99.0 | 1.1343 | .1051 |
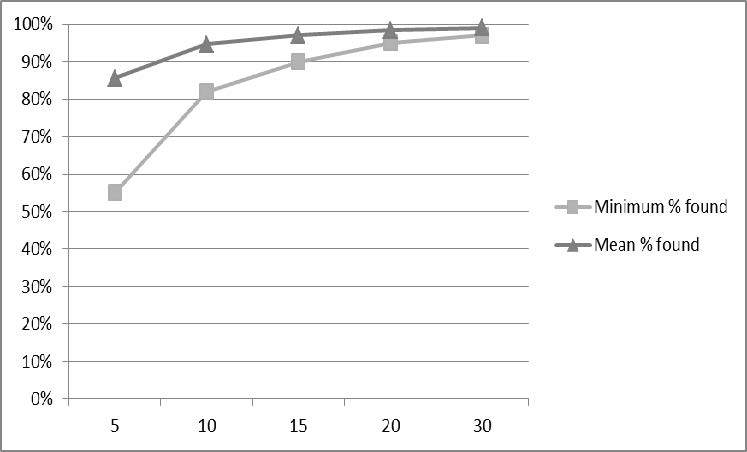
Human factors validation testing is primarily a qualitative rather than a quantitative exercise. The goal is to evaluate users’ interactions with a device user interface by observing their performance and simultaneously collecting subjective user assessments of their experience using the device to assess the adequacy of the user interface design. Use errors are recorded but the purpose is not to quantify the frequency of any particular use error or establish acceptability with respect to numerical acceptance criteria. Instead, the purpose is to identify the part of the user interface involved in a use error or problem and investigate the causes of the use error or problem so that the design of the user interface can be optimized with regard to use safety and effectiveness.
Since the parameters needed to determine sample size cannot be estimated easily or cannot be at estimated at all prior to testing, a sample of 15 people to detect most of the problems in a user interface constitutes a practical minimum number of participants for human factors validation testing. This sample size theoretically provides the best possibility of detecting user interface design flaws while limiting the amount of resources required. However, please note that the recommended minimum number of participants could be higher for specific device types.
Appendix C Analyzing Results of Human Factors Validation Testing 🔗
Analysis of human factors validation test results should focus on any problems found during the testing. Problems are use errors and “close calls” on critical tasks observed by the test facilitators (observational data) and difficulties with use, including close calls, reported by the test participants (interview data). If the testing was conducted adequately and no use errors or problems that could result in harm were found, the test data would require minimal analysis. More often problems are found and the test results require analysis to determine the severity of the potential harm that could result and if the harm could be serious, require identification of the root causes.
For those use errors and problems that could result in serious harm, the test data should be analyzed to determine which part of the user interface was involved and how the user interaction could have resulted in the use error or problem. The primary purpose of the analysis is to determine whether that part of the user interface could and should be modified to reduce or eliminate the use problem and reduce the use-related risks to acceptable levels. An essential secondary purpose of the analysis is to develop a modified design that would not cause the same problem or a new problem.
Even when the causes of the use errors and problems seem to be apparent from the test facilitators’ observations, they should always be discussed as part of the post-use interview. The test participant’s perspective on use errors can provide helpful insights and reasons for the use error and sometimes includes suggestions for design improvements. It is not uncommon for the user to explain exactly what caused them to do what they did but this is not always the case. Sometimes users don’t notice making errors, or cannot provide an explanation, or offer an explanation that is not helpful.
Design modifications made in response to human factors validation testing results to eliminate or reduce unacceptable use-related risks should be evaluated in a subsequent test to determine whether the design modifications were effective and whether they have introduced unacceptable new risks that need to be eliminated or reduced.
A hypothetical example: Three participants in a human factors validation study initiated purge of an infusion set prior to disconnecting the line from the patient. The use errors were observed but it was not clear why they happened. Subsequent discussion with the test participants revealed that they were confused by the appearance of the purge options shown on the device’s graphic user interface (GUI). In addition, two other test participants mentioned the same problem although they did not make the error; all five participants offered suggestions to make the user interface easier to understand. Analysis of the test results indicated clearly that the display screen for this function should be modified and also revealed possible ways in which the GUI could be improved. The user interface was revised and when the modified device was re-tested, the participants made no use errors on this task, did not report confusion or difficulties with it, and no new use problems were found.
Some use errors cannot be eliminated completely. For instance, despite specific instruction and warning that users should use disinfectant wipe prior to lancing a finger (or other site) to draw blood for a blood glucose test, several test participants omitted this step during a human factors validation test. Data collected during the post-task interviews indicated that the participants were aware of the risk of infection and read the warning in the instructions; however, they chose not to use the disinfectant wipe because, they said, “that’s just not how I do it.” These types of use errors should be discussed in the context of the risk control measures applied (e.g., clear information in the blood glucose meter’s user manual with validated cleaning and disinfection procedures using EPA-registered disinfecting products). Because further modification of the user interface would not be likely to reduce the use error rate and because the benefits of using the meter outweigh the risk of infection resulting from the use error, the residual risk would be acceptable.
Finally, some use errors that occur during a human factors validation test are found, through interviews with the test participants, to have been caused by conditions that were not consistent with actual use. Once so determined, such errors may be designated “test artifacts” and this conclusion is acceptable. However, analyses of test results that include many “artifact” explanations might indicate that the test conditions affected participant behavior too significantly and retesting might be necessary under conditions that more closely approximate real-world use. Some hypothetical examples of the process and results of analyzing human factors validation test results are shown in Table C.1 below.
Table C.1 Hypothetical sample results of analyses of human factors validation tests
Example 1 🔗
Medical device use task:
Enter patient data into medical device.
Hypothetical task failure (observations of user):
- User who was interrupted during the task failed to enter data into some fields.
- Users in US entered the patient weights in pounds rather than in kilograms.
Hypothetical task failure (comments by user):
- User was not aware he left some fields blank.
- Users did not realize that the medical device only recorded patient weights in kilograms.
Initial risk analysis (clinical consequences):
Incomplete patient data could lead to misdiagnosis
Initial risk analysis (potential harm):
Serious injury or death
Possible root cause
- User was not informed they left some fields blank.
- User could not tell the medical device accepted patient weights only in kilograms.
Possible risk control
- Medical device to alert the user when data has not been entered into required fields.
- Medical device to make the weight units more noticeable.
Evaluation of risk control effectiveness
HF validation test
Revised risk anal: redesign needed?
Example 2 🔗
Medical device use task:
Confirm new settings entered into a medical device.
Hypothetical task failure (observations of user):
- User did not confirm the settings.
- User did not notice the medical device did not keep the settings.
Hypothetical task failure (comments by user):
- User did not realize she needed to confirm the settings.
- User presumed the medical device kept the settings he had entered.
Initial risk analysis (clinical consequences):
Incorrect therapy
Initial risk analysis (potential harm):
Serious injury or death
Possible root cause
- User was not informed of the need to confirm the settings.
- User was not informed that the medical device had reverted to the previous settings.
Possible risk control
- Medical device to alert the user to confirm the settings.
- Medical device to alert the user when it has timed out and reverted to the previous settings.
Evaluation of risk control effectiveness
HF validation test
Revised risk anal: redesign needed?
Example 3 🔗
Medical device use task:
Read the medical device display and determine the status of the patient.
Hypothetical task failure (observations of user):
User misread the medical device display.
Hypothetical task failure (comments by user):
The medical device display was difficult to read.
Initial risk analysis (clinical consequences):
Incorrect therapy
Initial risk analysis (potential harm):
Serious injury or death
Possible root cause
The medical device display was difficult to read.
Possible risk control
- Increase the contrast between the display background and the text.
- Increase the size of the font used for critical information.
- Use non-glare glass on the medical device display.
Evaluation of risk control effectiveness
HF validation test
Revised risk anal: redesign needed?
Example 4 🔗
Medical device use task:
Respond to an auditory alarm signal coming from a medical device in the next room.
Hypothetical task failure (observations of user):
Users did not respond to the alarm signal.
Hypothetical task failure (comments by user):
Users could not hear the alarm signal.
Initial risk analysis (clinical consequences):
Alarm condition not addressed
Initial risk analysis (potential harm):
Serious injury or death
Possible root cause
- The frequency of the alarm signal was too high for some users to hear.
- The alarm tone was not loud enough for some users to hear.
Possible risk control
- Use alarm tones with multiple frequency components.
- Communicate with the user using a distributed alarm system that does not require hearing.
Evaluation of risk control effectiveness
HF validation test
Revised risk anal: redesign needed?
Example 5 🔗
Medical device use task:
Pause the alarm signal temporarily.
Hypothetical task failure (observations of user):
User permanently inactivated the alarm signals.
Hypothetical task failure (comments by user):
User thought he was pausing the alarm signal.
Initial risk analysis (clinical consequences):
Alarm condition might not be not addressed
Initial risk analysis (potential harm):
Serious injury or death
Possible root cause
In spite of the text “silence” appearing adjacent to the symbol, the user confused the alarm-pause control with the alarm-off control.
Possible risk control
- Follow IEC 60601-18 and do not use the historical term “silence,” which has had different meanings on different equipment.
- Add a confirmation step on the user interface for permanently inactivating the alarm signals of an alarm condition.
Evaluation of risk control effectiveness
HF validation test
Revised risk anal: redesign needed?
Example 6 🔗
Medical device use task:
Perform the procedure.
Hypothetical task failure (observations of user):
User did not respond to a warning that the medical device was overheating.
Hypothetical task failure (comments by user):
User didn’t believe the medical device warning because of repeated false alarms on the device.
Initial risk analysis (clinical consequences):
The medical device overheated and was inoperable, causing delay of therapy or absence of therapy.
Initial risk analysis (potential harm):
Serious injury or death
Possible root cause
User misinterpreted a valid alarm for a false alarm
Possible risk control
- Reduce the occurrence of false alarms associated with this problem.
- Emphasize warning in user manual.
- Address issue with overheating and alarm in training.
Evaluation of risk control effectiveness
HF validation test
Revised risk anal: redesign needed?
Example 7 🔗
Medical device use task:
Connect the components.
Hypothetical task failure (observations of user):
User broke the connector.
Hypothetical task failure (comments by user):
User couldn’t tell when the connection was secure so he over tightened it.
Initial risk analysis (clinical consequences):
Delay of therapy or absence of therapy
Initial risk analysis (potential harm):
Serious injury or death
Possible root cause
- Connector is not strong enough.
- Connector does not provide feedback to user when it is secure.
Possible risk control
- Redesign the connector to withstand greater torque.
- Redesign the connector to provide a snap sound and feel when it is secure.
Evaluation of risk control effectiveness
HF validation test
Revised risk anal: redesign needed?
Example 7 🔗
Medical device use task:
Set up the hemodialysis equipment.
Hypothetical task failure (observations of user):
User connected the fresh dialysate and the used dialysate to the opposite ports.
Hypothetical task failure (comments by user):
The two dialysate containers looked very similar.
Initial risk analysis (clinical consequences):
Inadequate therapy or toxic therapy
Initial risk analysis (potential harm):
Serious injury or death
Possible root cause
- The connectors on the two device ports and two dialysate containers were identical.
- The different dialysate containers were not visually distinctive.
Possible risk control
- Use different connectors on the two device ports and two dialysate containers so a wrong connection is not possible.
- Redesign the labels on the dialysate containers to be more distinctive.
Evaluation of risk control effectiveness
- None
- HF validation test
Revised risk anal: redesign needed?
Example 8 🔗
Medical device use task:
Start the therapy.
Hypothetical task failure (observations of user):
User pressed the “Enter” button rather than the “Start” button.
Hypothetical task failure (comments by user):
User presumed that the “Enter” button would start the therapy.
Initial risk analysis (clinical consequences):
Delay of therapy or absence of therapy
Initial risk analysis (potential harm):
Serious injury or death
Possible root cause
User did not understand the sequence of medical device operation.
Possible risk control
Medical device to add prompts to the display to remind the user to press “Start” to start the therapy.
Evaluation of risk control effectiveness
HF validation test
Revised risk anal: redesign needed?
Example 9 🔗
Medical device use task:
Suction fluid from the patient’s body cavity.
Hypothetical task failure (observations of user):
User connected the low-suction medical device to a high-suction vacuum source.
Hypothetical task failure (comments by user):
User did not realize that the medical device was not supposed to be connected to a high-suction vacuum source.
Initial risk analysis (clinical consequences):
Extraction of body tissues
Initial risk analysis (potential harm):
Serious injury or death
Possible root cause
User did not know they should not connect the medical device to a high-suction vacuum source.
Possible risk control
- Revise the labels on the medical device.
- Revise the instructions for use.
- Revise the user training.
Evaluation of risk control effectiveness
HF validation test
Revised risk anal: redesign needed?
Example 10 🔗
Medical device use task:
Check the expiration date on the component.
Hypothetical task failure (observations of user):
User did not check the expiration date.
Hypothetical task failure (comments by user):
User did not expect that the components used in the test might have expired.
Initial risk analysis (clinical consequences):
Inadequate therapy
Initial risk analysis (potential harm):
Serious injury
Possible root cause
Test artifact
Possible risk control
None
Evaluation of risk control effectiveness
None
Revised risk anal: redesign needed?
No
Example 11 🔗
Medical device use task:
Replace the (dead) battery.
Hypothetical task failure (observations of user):
User was not able to open the battery compartment door.
Hypothetical task failure (comments by user):
The battery door was too hard to open.
Initial risk analysis (clinical consequences):
Delay of therapy or absence of therapy
Initial risk analysis (potential harm):
Serious injury or death
Possible root cause
Opening the battery compartment door required more force than some users could generate.
Possible risk control
Redesign the battery compartment door to require less force and dexterity to open.
Evaluation of risk control effectiveness
HF validation test
Revised risk anal: redesign needed?
Appendix D - HFE/UE References 🔗
D.1 FDA Advice and Guidance Documents
To facilitate premarket review and assist manufacturers, FDA has published advice as well as device-specific and general guidance documents. As of this writing, FDA advice and guidance documents relevant to human factors are:
- Human Factors Implications of the New GMP Rule Overall Requirements of the New Quality System Regulation,
- Design Control Guidance for Medical Device Manufacturers,
- Total Product Life Cycle: Infusion Pump - Premarket Notification [510(k)] Submissions, and
- Design Considerations for Devices Intended for Home Use.
D.2 National and International Standards
FDA has officially recognized device-specific and general consensus standards published by national and international standards bodies. Standards recognized by FDA as of this writing related to human factors and the application of HFE/UE to medical devices are listed in Table D-1. Please note that the currently-recognized standards are noted at http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfStandards/search.cfm. It is important to check this page to review the supplementary information sheets (SIS) for all recognized standards to understand the extent of Agency recognition of each standard.
Table D-1. National and international consensus standards involving HFE/UE and recognized by FDA.
| Standard | Title | Main Purpose |
|---|---|---|
| AAMI/ANSI HE75 | Human Factors Engineering – Design of Medical Devices | Comprehensive reference that includes general principles, , management of use error risk, design elements, integrated solutions |
| ANSI/AAMI/IEC 62366 | Medical devices – Application of usability engineering to medical devices | HFE/UE process applied to all applying HF/usability to medical device design, with consideration of risk management |
| ANSI/AAMI/ISO 14971 | Medical Devices – Application of risk management to medical devices | Risk management process for medical devices |
| IEC 60601-1-6 | Medical electrical equipment – Part 1-6: General requirements for basic safety and essential performance – Collateral standard: Usability |
Provides a bridge between IEC 60601-1 and ANSI/AAMI/IEC 62366 |
| IEC 60601-1-8 | Medical electrical equipment — Part 18: General requirements for basic safety and essential performance — Collateral Standard: General requirements, tests and guidance for alarm systems in medical electrical equipment and medical electrical systems | Design standard for alarm systems in medical electrical equipment and systems |
| IEC 60601-1-11 | Medical electrical equipment – Part 1-11: General requirements for basic safety and essential performance – Collateral Standard: Requirements for medical electrical equipment and medical electrical systems used in the home healthcare environment | Requirements for medical electrical equipment used in non-clinical environments, including issues involving medical device use by lay users |
D.3 Additional HFE/UE References
Dumas, J. and Loring, B. (2008). Moderating Usability Tests: Principles and Practices for Interacting. San Francisco, CA: Elsevier/Morgan Kauffman Publishers.
Faulkner, L. (2003). Beyond the five-user assumption: Benefits of increased sample sizes in usability testing. Behavior Research Methods, Instruments, and Computers, 35(3), 379-383.
Hackos, J. and Redish, J. (1998). User and Task Analysis for User Interface Design. New York: John Wiley & Sons.
Israelski, E.W. and Muto, W.H. (2006). Risk management in medical devices. In: Carayon P (Ed.). Handbook of human factors and ergonomics in health care and patient safety. Philadelphia (PA): Lawrence Erlbaum Associates.
Kaye, R. D, North, R.A., and Peterson, M. K. (2003) UPCARE: An analysis, description, and educational tool for medical device use problems. Proceedings of the 9th Annual International Conference on Industrial Engineering Theory, Applications and Practice. Las Vegas, NV.
Kirwan, B., and Ainsworth, L.K. (1992). A Guide to Task Analysis. London: Taylor & Francis Ltd;
Meister, D. (1986). Human factors testing and evaluation. Amsterdam: Elsevier.
Morrow D, North RA, and Wickens CD. Reducing and mitigating human error in medicine. In: Nickerson R (Ed.). Reviews of human factors and ergonomics. Vol. 1. Santa Monica (CA):, Human Factors and Ergonomics Society, 2006.
Nielsen, J. (1993). Usability Engineering. Boston: AP Professional.
Norman, D., The Design of Everyday Things. New York: Doubleday; 1988.
Reason, J., Human Error. Cambridge, Mass: Cambridge University Press; 1992.
Rubin, J. and Chisnell, D., (2008). Handbook of Usability Testing: How to Plan, Design, and Conduct Effective Tests. New York: John Wiley and Sons, Inc.
Salvendy, G. (ed.), Handbook of Human Factors and Ergonomics. New York: John Wiley and Sons, Inc; 1997.
Sanders, M., and McCormick E., Human Factors in Engineering and Design. New York: McGraw Hill; 1993.
Shneiderman, B., Plaisant, C., Cohen, M. and Jacobs, S. (2010). Designing the User Interface: Strategies for Effective Human-Computer Interaction. (5th ed.). Menlo Park, CA: Addison Wesley.
Story, M.F. (2010). Medical Devices in Home Health Care. In National Research Council, The Role of Human Factors in Home Health Care: Workshop Summary, Olson, S, Rapporteur. Committee on Human-Systems Integration, Division of Behavioral and Social Sciences and Education. Washington, DC: The National Academies Press.
Trautman, K. (1997). The FDA and Worldwide Quality Systems Requirements Guidebook for Medical Devices. ASQC Press.
Usability.gov (2013). Heuristic Evaluations and Expert Reviews. Retrieved October 20, 2014, from Usability.gov, How To & Tools: http://www.usability.gov/how-to-andtools/methods/heuristic-evaluation.html
Virzi, R.A. (1992). Refining the rest phase of usability evaluation: How many subjects is enough? Human Factors, 34, 457-468.
Wiklund, M.E., Kendler, J. and Strochlic, A.Y. (2011). Usability Testing of Medical Devices. Boca Raton, FL: Taylor & Francis/CRC Press.
Footnotes 🔗
-
21 CFR 820.30 ↩
-
ANSI/AAMI/ISO 14971:2007, definition 2.16 ↩
-
In the US, the term “human factors engineering” is predominant but in other parts of the world, “usability engineering” is preferred. For the purposes of this document, the two terms are considered interchangeable. ↩
-
Human factors validation testing is sometimes referred to as “summative usability testing.” However, summative usability testing can be defined differently and some definitions omit essential components of human factors validation testing as described in this guidance document. ↩
-
Actual-use clinical studies conducted in the United States must comply with the Investigational Device Exemption requirements set out in 21 CFR §812. ↩
-
e.g., Virzi, 1992; Nielsen, 1993 ↩
