
Innolitics introduction 🔗
This FDA guidance is important if you’re building a device that detects abnormalities in radiological images or data (CADe). Even if your device is not CADe, many of the performance assessment study design principles and documentation discussed in this guidance can be useful. In particular, we’ve had the FDA refer to this guidance even for product codes note listed as in scope.
Designing standalone and clinical performance testing studies for AI/ML-enabled SaMD can be tricky. You want to do enough testing to ensure the device is safe and effective, yet you also need to avoid going bankrupt on the way! Sourcing and annotating data can be costly. We have a lot of experience helping companies design practical studies using our combined understanding of the regulatory considerations and AI/ML design. If you could use help, please reach out in the chat or on our contact page.
About this Transcript 🔗
This document is a transcript of an official FDA (or IMDRF) guidance document. We transcribe the official PDFs into HTML so that we can share links to particular sections of the guidance when communicating internally and with our clients. We do our best to be accurate and have a thorough review process, but occasionally mistakes slip through. If you notice a typo, please email a screenshot of it to Miljana Antic at mantic@innolitics.com so we can fix it.
Preamble 🔗
Document issued on September 28, 2022.
Document originally issued on July 3, 2012.
For questions about this document, contact RadHealth@fda.hhs.gov.
Contains non-binding guidance.
I. Introduction 🔗
This guidance document provides recommendations to industry, systems and service providers, consultants, FDA staff, and others regarding premarket notification (510(k)) submissions for computer-assisted detection (CADe1) devices applied to radiology images and radiology device data (often referred to as “radiological data” in this document). CADe devices are computerized systems that incorporate pattern recognition and data analysis capabilities (i.e., combine values, measurements, or features extracted from the patient radiological data) and are intended to identify, mark, highlight, or in any other manner direct attention to portions of an image, or aspects of radiology device data, that may reveal abnormalities during interpretation of patient radiology images or patient radiology device data by the intended user (i.e., a physician or other health care professional), referred to as the “clinician” in this document. We have considered the recommendations on documentation and performance testing for CADe devices made during the Radiology Devices Panel meetings on March 4-5, 2008,22 and November 17-18, 2009.33 We have also considered the public comments received on the draft guidance announced in the Federal Register on October 21, 2009 (74 FR 54053).4
In general, FDA’s guidance documents do not establish legally enforceable responsibilities. Instead, guidances describe the Agency’s current thinking on a topic and should be viewed only as recommendations, unless specific regulatory or statutory requirements are cited. The use of the word should in Agency guidances means that something is suggested or recommended, but not required.
II. Background 🔗
This guidance applies to the CADe devices identified in Section III. Scope by their classification regulation (21 CFR 892.2050) and product codes (NWE, OEB, OMJ). A manufacturer who intends to market one of these devices must:
- conform to the general controls of the Federal Food, Drug, and Cosmetic Act (FD&C Act), including the 510(k) requirements described in 21 CFR 807 Subpart E;
- conform to the special controls designated for this device (see 21 CFR 892.2050(b)); and
- obtain a substantial equivalence determination from FDA prior to marketing the device (see 21 CFR 807 Subpart E).
This document provides recommendations regarding 510(k)s for these devices. It supplements the requirements in 21 CFR 807 Subpart E, and recommendations in other FDA documents concerning the specific content of a 510(k) submission, including the guidance “Format for Traditional and Abbreviated 510(k)s.”5
This guidance was revised with minor updates to reflect the issuance of the final rule, “Medical Devices; Medical Device Classification Regulations To Conform to Medical Software Provisions in the 21st Century Cures Act” (86 FR 20278).6
III. Scope 🔗
This document provides guidance regarding 510(k) submissions for CADe devices applied to radiology images and radiology device data. Radiological data include, for example, images that are produced during patient examination with ultrasound, radiography, magnetic resonance imaging (MRI), computed tomography (CT), positron emission tomography (PET), and digitized film images. As stated above, CADe devices are computerized systems intended to identify, mark, highlight, or in any other manner, direct attention to portions of an image, or aspects of radiology device data, that may reveal specific abnormalities during interpretation of patient radiology images or patient radiology device data by the clinician. This guidance covers CADe devices marketed as a complete package with a review workstation, or as an add-on software to be embedded within imaging equipment, an image review platform (for example, a medical image management and processing system), or other imaging accessory equipment.
This guidance document applies to the CADe devices classified under 21 CFR 892.2050 medical image management and processing systems, including the following product codes:
- NWE (Colon computed tomography system, computer-aided detection);
- OEB (Lung computed tomography system, computer-aided detection); and
- OMJ (Chest x-ray, computer-aided detection).
The Agency intends to create new product codes to identify and track new types of CADe products as necessary.7
This guidance does not apply to (1) clinical performance assessment studies for CADe devices that are intended for use during intra-operative procedures, (2) radiological computer-assisted diagnostic (CADx) devices (classified under 21 CFR 892.2060), (3) computer-assisted detection/diagnosis (CADe/x) devices (classified under 21 CFR 892.2090), (4) computer-assisted acquisition/optimization (CADa/o) devices (classified under 21 CFR 892.2100), or (5) radiological computer-assisted triage and notification (CADt) devices (classified under 21 CFR 892.2080), whether marketed as unique devices or bundled with a CADe device that, by itself, may be subject to this guidance. The guidance also does not apply to the many non-CADe devices that are also classified under 21 CFR 892.2050, including, for example, product codes LLZ (System, Image Processing, Radiological) and NFJ (System, Image Management, Ophthalmic). Therefore, most basic radiological image processing and image acquisition products fall outside the scope of this guidance document.
21 CFR 892.2050 Medical image management and processing system.
- Identification. A medical image management and processing system is a device that provides one or more capabilities relating to the review and digital processing of medical images for the purposes of interpretation by a trained practitioner of disease detection, diagnosis, or patient management. The software components may provide advanced or complex image processing functions for image manipulation, enhancement, or quantification that are intended for use in the interpretation and analysis of medical images. Advanced image manipulation functions may include image segmentation, multimodality image registration, or 3D visualization. Complex quantitative functions may include semi-automated measurements or time-series measurements.
- Classification. Class II (special controls; voluntary standards – Digital Imaging and Communications in Medicine (DICOM) Std., Joint Photographic Experts Group (JPEG) Std., Society of Motion Picture and Television Engineers (SMPTE) Test Pattern).
By design, a CADe device can be a unique detection scheme specific to only one type of potential abnormality or a combination or bundle of multiple parallel detection schemes, each specifically designed to detect one type of potential abnormality that is revealed in the patient radiological data.
Examples of CADe devices that fall within the scope of this guidance include:
- a CADe device intended to identify and prompt colonic polyps on CT colonography studies;
- a CADe device intended to identify and prompt filling defects on thoracic CT examination;
- a CADe device intended to automatically detect liver lesions; and
- a CADe device intended to identify lung nodules on CT, x-ray, or MRI studies.
Examples of devices that do not fall within the scope of this guidance include:
- software intended to provide 3D visualization of the colon;
- software intended to segment and visualize the airway tree;
- software intended to segment the liver;
- software intended to measure the size of a pre-identified tumor; and
- software intended to identify lymph nodes.
This guidance does not cover any CADe devices that are intended for use during intra-operative procedures, or any computer-assisted diagnostic devices (CADx) or computer-triage devices, whether marketed as unique devices or bundled with a computer-assisted detection device that, by itself, may be subject to this guidance. Below is further explanation of the CADx and computer triage devices not covered by this guidance:
- CADx devices are computerized systems intended to provide information beyond identifying, marking, highlighting, or in any other manner directing attention to portions of an image, or aspects of radiology device data, that may reveal abnormalities during interpretation of patient radiology images or patient radiology device data by the clinician. CADx devices include those that are intended to provide an assessment of disease or other conditions in terms of the likelihood of the presence or absence of disease, or are intended to specify disease type (i.e., specific diagnosis or differential diagnosis), severity, stage, or intervention recommended. An example of such a device would be a computer algorithm designed both to identify and prompt lung nodules on CT exams and also to provide a probability of malignancy score to the clinician for each potential lesion as additional information.
- Computer-triage devices are computerized systems intended to, in any way, reduce or eliminate any aspect of clinical care currently provided by a clinician, such as a device for which the output indicates that a subset of patients (i.e., one or more patients in the target population) are normal and therefore do not require interpretation of their radiological data by a clinician. An example of this device is a prescreening computer scheme that identifies patients with normal MRI scans that do not require any review or diagnostic interpretation by a clinician.
For any of these types of devices, we recommend that you contact the Agency to inquire about premarket pathways, regulatory requirements, and recommendations about non-clinical and clinical data.
IV. Describing the Device in a 510(k) 🔗
We recommend you identify your device by the regulation and product code described in Section III. Scope, if applicable, and provide an overview of your CADe algorithm and a description of the following:
- the algorithm design and function,
- processing steps,
- features,
- models and classifiers,
- training paradigm,
- databases,
- reference standard, and
- scoring methodology.
A. General Information 🔗
In providing a description of your device, we recommend that you include the following information:
- target population information including patient population, organs of interest, diseases/conditions/abnormalities of interest, and appropriate clinician intended to use the device (e.g., radiologist, family practice physician, nurse);
- radiological data used as an input to and compatible with your CADe design, including imaging modalities (e.g., computed tomography, magnetic resonance), make, model and specific trade name for each modality/system, if applicable, specific image acquisition parameter ranges (e.g., kVp range, slice thickness), and specific clinical imaging protocol(s) (e.g., oral contrast studies, magnetic resonance imaging (MRI) sequence);
- current clinical practice relevant to the diseases/conditions/abnormalities of interest;
- proposed clinical workflow (as compared to the predicate device) including a description of:
- how your device is labeled for use in clinical practice,
- when your device should be utilized within the proposed workflow, and
- effects on interpretation time when specific claims are made;
- device impact (as compared to the predicate device), including:
- the impact on the patient associated with device performance for both true positive and true negative marks, and
- the impact on the patient associated with device performance for false positive and false negative marks, separately (e.g., an incorrect follow-up recommendation based on a false positive detection would likely result in short term surveillance imaging for the patient);
- device limitations (as compared to the predicate device), including diseases/conditions/abnormalities for which the device has been found ineffective and should not be used; and
- supporting data from the scientific literature.
B. Algorithm Design and Function 🔗
This guidance recommends that you include a detailed description of your CADe algorithm in your 510(k). There may be instances where a general description of the algorithm and its stages are enough to provide an appropriate level of understanding for Agency review, especially when a clinical performance assessment is included as part of your performance testing. However, there may also be instances, especially when comparing a modified CADe device with a previously marketed CADe device, where a more detailed description of both original and modified CADe devices may reduce the performance testing requirements that may be needed to evaluate the modified device. The Agency recommends that the level of detail be at the level to allow for a technological comparison with any predicate devices.
We recommend that you provide information on the algorithm design and function including details on algorithm implementation that provides a description of the format of all CADe marks available, including all relevant geometric and other properties such as shape, size, intended location in relation to region of interest (e.g., overlap, adjacent), border (e.g., solid, dashed), and color. For CADe devices designed as add-on software for image review workstations and medical image management systems, the Agency recommends developing requirements for appropriately displaying the CADe marks and including these as part of your submission.
We recommend that you provide a flowchart identifying the processing, features, models, and classifiers utilized by your algorithm. We suggest that your flowchart include the following:
- all manual operations and associated predefined default settings (e.g., selection of rules or thresholds by the physician);
- all semi-automatic operations and associated predefined default settings (e.g., selection of seed points for region segmentation); and
- all automatic operations that do not involve direct interaction with the clinician.
You should also include other algorithm/software information including:
- name,
- version, and
- relevant characteristics of the software platform.
We recommend that you briefly describe the design and function for each stage of your algorithm, where a stage is an independent or well-defined functional unit within the CADe algorithm. The description should provide a basic understanding of each stage of the CADe detection process. If your technique is similar to that of a published study, a reference to that study, and/or inclusion of the article, is encouraged. Your description should include a discussion of the following:
- purpose of the stage,
- processing steps,
- features,
- models and classifiers,
- training paradigm,
- development and training databases utilized, and
- reference standard.
We also recommend that you tabulate and describe the similarities and differences in the technological characteristics of your devices with that of the predicate devices to facilitate the review process.
C. Processing 🔗
Processing refers to any image or signal processing steps included in the CADe algorithm. Examples of processing include filtering (e.g., a noise reduction filter), segmentation of areas or structures of interest (e.g., delineation of an organ of interest from its surroundings), normalization, registration, and artifact or motion correction. We recommend that you provide a description of all processing as well as relevant algorithm flowcharts and references.
Normalization processing refers to calibration or transformation of image or signal characteristics to that of a reference image or signal. We recommend that you provide a description of the technique used to establish the proper calibration or transformation, as well as the characteristics of the reference.
D. Features 🔗
Features are computer or human estimated quantities characterizing images, regions, or pixels within radiological data, including specific patient characteristics (e.g., age, sex, race, ethnicity). For each stage of your algorithm, we recommend that you provide:
- the feature classes used by the algorithm (e.g., demographic, biological, morphological, and geometrical features);
- a brief description of the features included as part of the commercial product; and
- a brief description of any feature selection (i.e., technique used to cull the set of candidate features) or dimensionality reduction (i.e., technique used to reduce the dimensionality of the feature space) processes.
E. Models and Classifiers 🔗
We define a model as any method or rule used to combine information. A classifier is a human or mathematically defined model used to rate or categorize regions within an image with respect to disease, condition, or abnormality. This model is an assumed relationship between image features and the rating or categorization of disease, condition, or abnormality, and depends on a specific set of parameters that are determined either manually or automatically. Models and classifiers typically perform some type of pattern recognition procedure. They can vary from a single threshold on a uniquely extracted feature to a complex classifier (i.e., a weighted combination of feature values). For each stage of your algorithm, we recommend that you provide a description of the types of models and classifiers used (e.g., simple threshold, decision tree, linear discriminant, neural network, support vector machine), the total number of input features utilized in each model or classifier, and the order in which they are applied.
F. Algorithm Training 🔗
Algorithm training is a procedure used to establish algorithm parameters and thresholds. This procedure includes the adjustment of filter parameters, the selection of the most discriminant features, and the adjustment of classifier weights and model parameters. Training may be done manually by humans (e.g., the programmer or a medical professional), automatically using a specialized training algorithm, or by a combination of both. We recommend that you provide a brief summary of your algorithm training paradigm and indicate if it is performed:
- manually by humans,
- automatically using a computerized training method, or
- by a combination of manual and computerized techniques.
We also recommend that you describe the criteria and performance metrics used to determine parameter settings and provide a summary of the incremental algorithm performance for appropriate intermediate stages of the algorithm.
G. Databases 🔗
Databases refer to the sets of radiology images or radiology device data used in training and testing your device (i.e., training and testing cases). These databases may contain computer simulated data, phantom data, or patient data depending on the nature of the evaluation.
For a database of computer simulated or phantom data, we recommend that you provide:
- a description of the phantom or simulation methodology; and
- any data characterizing the relationship between the simulated or phantom data and actual patient data for the imaging technique, organ, and disease of interest.
For each database of patient data (i.e., training and testing cases), we recommend that you provide specific information including (if available):
- the inclusion and exclusion criteria for patient data collection;
- the patient demographic data (e.g., age, ethnicity, race, sex);
- the patient medical history relevant to the CADe application;
- the patient disease state and indications for the radiologic test;
- the conditions of radiologic testing, for example, technique (including whether the test was performed with/without contrast, contrast type and dose per patient, patient body mass index if relevant, radiation exposure, and T1-weighting for MRI images) and views taken;
- a description of how the imaging data were collected (e.g., make and model of imaging devices and the imaging protocol);
- the collection sites;
- the processing sites, if applicable (e.g., patient data digitization);
- the number of cases, including:
- the number of diseased cases,
- the number of normal cases, and
- any methods used to determine disease status, location, and extent (see Section IV.H. Reference Standard);
- the case distributions stratified by relevant confounders or effect modifiers, such as lesion type (e.g., hyperplastic vs. adenomatous colonic polyps), lesion size, lesion location, disease stage, organ characteristics, concomitant diseases, imaging hardware (e.g., makes and models), imaging or scanning protocols, collection sites, and processing sites (if applicable);
- a comparison of the clinical, imaging, and pathologic characteristics of the patient data compared to the target population; and
- a history of the accrual and use of both training and test databases.
For CADe devices intended to be used with proprietary imaging devices, we recommend that you provide the trade names, regulatory status, and physical characteristics of these proprietary imaging devices.
H. Reference Standard 🔗
For purposes of this document, the reference standard (also often called the “gold standard” or “ground truth” in the imaging community) for patient data indicates whether or not the disease/condition/abnormality is present and may include such attributes as the extent or location of the disease/condition/abnormality. CADe device development and evaluation often relies on databases of radiology images or radiology device data with a reference standard addressing whether or not the disease/condition/abnormality is present within an individual patient and if so, its location and extent. We refer to this characterization of the reference standard for the patient, e.g., disease status, as the truthing process.
The methodology utilized to establish the reference standard can impact reported performance. You should provide the rationale and describe the procedure for defining if a disease/condition/abnormality is present and the location and extent of the disease/condition/abnormality (e.g., based on pathology or based on a standard of care determination). You should also indicate if the reference standard is based on:
- the output from another device;
- an established clinical determination (e.g., biopsy, specific laboratory test);
- a follow-up clinical imaging examination;
- a follow-up medical examination other than imaging; or
- an interpretation by reviewing clinician(s) (i.e., clinical truther(s)).
The methodology utilized to make this reference standard determination should be described and should be fixed prior to initiating your evaluation. For truthing that relies on the interpretation by reviewing clinician, we recommend that you provide:
- the number of clinical truthers involved;
- their qualifications;
- their levels of experience and expertise;
- the instructions conveyed to them prior to participating in the truthing process;
- all available clinical information from the patient utilized by them in the identification of disease/condition/abnormality and in the marking of the location and extent of the disease/condition/abnormality; and
- any specific criteria used as part of the truthing process.
When multiple clinical truthers are involved, you should describe the process by which their interpretations are combined to make an overall reference standard determination and how your process accounts for any inconsistencies between clinicians participating in the truthing process. Clinicians participating in the truthing process should not be the same as those who participate in the core clinical performance assessment of the CADe device because doing so would introduce bias into the study results.
I. Scoring 🔗
In addition to determining the reference standard for the location and extent of the disease/condition/abnormality, CADe device development and evaluation often rely on determining whether the spatial location and extent of a CADe mark correspond to the location and extent of the disease/condition/abnormality. We define the procedure for determining the correspondence between the CADe output and the reference standard (e.g., disease location) as the scoring process. The scoring procedure and the scoring definition are important components for interpreting standalone device performance and for appropriately labeling the device.
In this guidance, we describe the scoring used to evaluate device standalone performance. A different type of scoring is used in the clinical performance assessment, which is described in the FDA guidance entitled “Clinical Performance Assessment: Considerations for Computer-Assisted Detection Devices Applied to Radiology Images and Radiology Device Data in Premarket Notification (510(k)) Submissions.”8
Common scoring criteria that have been used to determine the nature of each CADe detection include:
- centroid of the CADe detection area or volume falling in the reference standard area or volume;
- distance between centroids of the CADe detection and the reference standard;
- ratio of the distance between centroids of the CADe detection and the reference standard, relative to the maximum width of the reference standard region;
- ratio of the area (A) or volume (V) intersection between the CADe detection and the reference standard, with the total area or volume of the CADe detection, defined as follows:
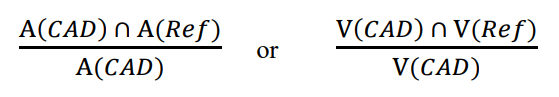
- ratio of the area (A) or volume (V) intersection between the CADe detection and the reference standard with the total area or volume union of the reference standard and the CADe detection, defined as follows:
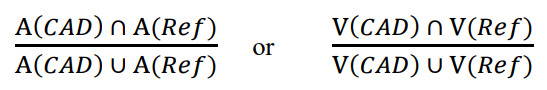
The scoring process should be consistent with the abnormalities being marked by the CADe and the intended use of your device and the performance claims being made. You should provide a scientific justification for this process and it should be fixed prior to initiating your evaluation. In your description of the scoring process, we recommend that you indicate whether the scoring is based on:
- electronic or non-electronic means;
- physical overlap of the boundary, area, or volume of the mark in relation to the boundary, area, or volume of the reference standard;
- relationship of the centroid of the mark to the boundary or spatial location of the reference standard;
- relationship of the centroid of the reference standard to the boundary or spatial location of the mark;
- interpretation by reviewing readers; or
- other methods.
For scoring that relies on interpretations by reviewing readers, we recommend that you provide the number of readers involved, their qualifications, their level of experience and expertise, the specific instructions conveyed to them prior to participating in the scoring process, and any specific criteria used as part of the scoring process. When multiple readers are involved in scoring, you should describe the process by which their interpretations are combined to make an overall scoring determination or how their interpretations are incorporated in the performance evaluation, including how any inconsistencies are addressed.
J. Other Information 🔗
We recommend that you include software documentation following the “Guidance for the Content of Premarket Submissions for Software Contained in Medical Devices”9 and the FDA guidance “Off-the-Shelf Software Use in Medical Devices.”10 The kind of information we that recommend submitting is determined by the “level of concern,” which is related to the risks associated with a software failure. The level of concern for a device may be minor, moderate, or major. Based on prior CADe device submissions, the level of concern for a CADe system is generally moderate.
If the CADe system is an add-on software to be installed within a third party image review platform, we recommend that you also provide the names, version/model numbers, and characteristics of these third party platforms as well as a description of the file format of the CADe output that is generated by your device.
V. Standalone Performance Assessment 🔗
Because each new CADe device represents a new implementation of the CADe algorithm and software, FDA expects that each new CADe device (as well as software and other design, technology, or performance changes to an already cleared CADe device) may have different technological characteristics from the legally marketed predicate device, even while sharing the same intended use. We may consider a change or modification in a CADe algorithm to significantly affect the safety or effectiveness of the device, for the purposes of this guidance, if, for example, a change has been made to the image processing components, features, classification algorithms, training methods, training data sets, or algorithm parameters. Accordingly, under section 513(i)(1)(A) of the FD&C Act, determinations of substantial equivalence will rest on whether the information submitted, including appropriate clinical or scientific data, demonstrate that the new or changed device is as safe and effective as the legally marketed predicate device and does not raise different questions of safety and effectiveness than the predicate device. We encourage you to contact the Agency for advice on whether your proposed changes or modifications to an existing CADe device constitute significant changes or modifications that require a premarket notification.11
To support a substantial equivalence determination for a new CADe device, or for changes to an already cleared CADe device that could significantly affect the safety or effectiveness of the device, we recommend that you measure and report the performance of your CADe device by itself, in the absence of any interaction with a clinician (i.e., standalone performance assessment). These measurements estimate how well the CADe device, by itself, marks regions of known abnormalities and how well the CADe device avoids marking regions other than the abnormalities (e.g., normal organ and structures). Study endpoints should be selected to establish meaningful and statistically relevant performance for the device.
To support substantial equivalence, we recommend comparing the standalone performance of your CADe device to the standalone performance of the predicate device on the same dataset, if possible. Otherwise, the characteristics or makeup of the database used to assess standalone performance of your device should be comparable to the characteristics or makeup of the database reportedly used in assessing the predicate device.
The types and nature of the abnormalities marked or not marked by your CADe device should be consistent with the intended use of your device. To measure standalone performance, you should determine the true location of abnormalities through some well-described truthing process (see Section IV.H. Reference Standard). You should compare the location and extent of a CADe mark to the truthed location and extent of an abnormality using the established scoring process (see Section IV.I. Scoring). You should establish the reference standard definition, scoring process, and analysis methodology, including primary and secondary performance endpoints, prior to the collection of the standalone performance assessment data and analysis of these data. You should demonstrate any performance claims based on a covariate analysis through a prespecified analysis plan.
We recommend that you perform standalone testing in a way that will provide good estimates of performance stratified by important covariates, such as lesion type, size, or shape. This stratified standalone performance is useful in labeling by providing the end users with additional information to better interpret the CADe marks.
A. Study Population 🔗
We recommend that you assess and report your device standalone performance on testing data that is independent and sequestered from the data on which you developed and trained the CADe.
Your testing database should be representative of the target population (e.g., include diverse demographic groups) and the target disease, condition, or abnormality for which your device is intended. We recommend that you provide the protocol for your case collections. An acceptable approach for acquiring data that is representative of the intended use population is the collection of consecutive cases from each participating collection site that fall within the inclusion and outside the exclusion criteria. The full range of diseased/abnormal and normal cases should be sufficiently represented in the testing database.
Enrichment with diseased/abnormal cases is permissible for an efficient and least burdensome representative case dataset, but may affect standalone performance estimates (e.g., the performance estimates may not generalize to the intended use population). You may choose to enrich the study population with patient cases that contain imaging findings (or other image data) that are known to challenge clinicians, but that still fall within the device’s intended use population (i.e., stress testing). When you use stress testing, selective sampling from cohorts within the intended use population is appropriate. For example, a study population enriched with cases containing small colonic polyps may be appropriate when assessing a general CADe device designed to assist in detecting a wide range of polyp sizes. Enrichment with cases based on the CADe device performance on those cases would not be appropriate.
The study should contain a sufficient number of cases from important cohorts (e.g., subsets defined by clinically relevant confounders, effect modifiers, and concomitant diseases) such that you can obtain standalone performance estimates and confidence intervals for these individual subsets (e.g., performance estimates for different nodule size categories when evaluating a lung CADe device). Powering these subsets for statistical significance is not necessary unless you are making specific subset performance claims.
A good study design might include and report results for both an enriched data set containing relevant confounders as well as a set of consecutive cases from each participating collection site where the consecutive cases may better represent the standalone performance in clinical practice.
The sample size of the study should be large enough such that the study has adequate power to detect with statistical significance your proposed performance claims. If performance claims are proposed for individual subsets, then you should determine the sample sizes for these subsets accordingly to detect these claims with statistical significance. When you are making multiple performance claims, a pre-specified statistical adjustment for the testing of multiple subsets would be necessary.
As part of the device standalone performance assessment, you should describe the testing database (see Section IV.G. Databases). We recommend that your performance testing include detection accuracy testing and localization accuracy testing.
B. Test Data Reuse 🔗
Reusing test data (i.e., conducting multiple tests on the same data) may be problematic for interpreting standalone performance results. The Agency recognizes the difficulty in acquiring data for CADe assessment. The Agency also recognizes that the CADe algorithm may implicitly or explicitly become tuned to the test data if the same test set is used multiple times. With this issue in mind, the Agency encourages evaluation of new or modified CADe algorithms using newly acquired independent test cases (i.e, the Agency discourages repeated use of test data in evaluating device performance). If you are considering data reuse in the evaluation of your CADe device’s standalone performance, you should demonstrate that reusing any part of the test data does not introduce unreasonable bias into estimates of CADe performance and that test data integrity is maintained.
In the event that you would like the Agency to consider the reuse of any test data in your standalone evaluation, you should control the access of your staff to performance results for the test subpopulations and individual cases. It may therefore be necessary for you to set up a “firewall” to ensure those outside of the regulatory assessment team (e.g., algorithm developers) are completely insulated from knowledge of the radiology images and radiological data, and anything but the appropriate summary performance results. You should maintain test data integrity throughout the lifecycle of the product. This is analogous to data integrity involving clinical trial data monitoring committees and the use of a “firewall” to insulate personnel responsible for proposing interim protocol changes from knowledge of interim comparative results. For more information on data integrity for clinical trial data monitoring committees, refer to Section 4.4.1.4 of the FDA guidance entitled “Establishment and Operation of Clinical Trial Data Monitoring Committees.”12
To minimize the risk of tuning to the test data and to maintain data integrity, we recommend that you develop an audit trail and implement the following controls when you contemplate the reuse of any test data:
- you randomly select the data from a larger database that grows over time;
- you retire data acquired with outdated image acquisition protocols or equipment that no longer represents current practice;
- you place a small, fixed limit on the number of times a case can be used for assessment;
- you maintain a firewall such that data access is tightly controlled to ensure that nobody outside of the regulatory assessment team, especially anyone associated with algorithm development, has access to the data (i.e., only summary performance results are reported outside of the assessment team);
- you maintain a data access log to track each time the data is accessed including a record of who accessed the data, the test conditions, and the summary performance results.
The purposes of the audit trail include: (1) establishing that you defined the cases in the training and test sets appropriately such that data leakage between training and test sets did not occur; (2) ensuring that you fixed the new CADe algorithm in advance (i.e., before application to the test set); and (3) providing information concerning the extent to which you used the same test set or a subset thereof for testing other CADe algorithms or designs, including results reported to the Agency as well as non-reported results. The controls we are recommending are intended to substantially reduce the chance that you evaluate a new CADe algorithm in a subsequent study using the same test data that you used for a prior CADe algorithm.
If you reuse test data, you should report the test performance for relevant subsets in addition to the overall performance. These subsets include: (1) the portion of the test set for the new CADe algorithm that overlaps with previously used test sets, and (2) the portion of the test set that you have never used before. Since these subsets will be smaller in size compared to the overall test set for the new CADe algorithm, confidence intervals for the subsets will be wider. However, the trends for the mean performances would be helpful to indicate whether you have tuned the CADe system to previously used portions of the test set.
C. Detection Accuracy 🔗
We recommend that you estimate and report the CADe standalone performance following the scoring process (see Section IV.I. Scoring). The definition of a true positive (TP), true negative (TN), false positive (FP), and false negative (FN) CADe mark should be consistent with the intended use of the device. For example, if the device is intended to detect all abnormalities (e.g., benign and malignant), then a true positive CADe mark should be defined as “marking” any abnormalities. On the other hand, if a device is intended to detect only a subset of abnormalities (e.g., only those lesions with certain imaging features), then you should define a true or false CADe mark accordingly.
For a reference standard (e.g., disease type, location, and extent) that relies on the interpretation by reviewing readers, we recommend that you account for reader variability in the truthing process and for various consensus or agreement rules between expert readers, in the CADe standalone performance estimates. One method of accounting for variability in the reference standard is to resample the expert truthing panel. See Miller et al.13 for details on one approach.
We recommend that you report the overall lesion-based, patient-based, or other relevant measure of standalone sensitivity (e.g., probability the CADe algorithm correctly marks the location of an abnormality) at each operating point. You should report a relevant measure of the standalone false positive rate14 (e.g., the average number of false positive marks per case [FPs/case]) at each device operating point. You should report stratified analysis per relevant confounder or effect modifier as appropriate (e.g., lesion size, lesion type, lesion location, disease stage, imaging or scanning protocols, imaging or data characteristics). The standalone false positive rate should be derived from normal and abnormal patient data separately. If your device allows the clinician to select or manipulate the device operating point, we recommend that you provide the device performance for each selectable operating point or for the range of possible operating points. In addition, detection accuracy will likely depend upon the scoring criteria and scoring threshold used to determine the nature and extent of each CADe detection (see Section IV.I. Scoring). Therefore, we also recommend that you estimate and report the performance of your device using various values of the distance and ratio scoring thresholds when applicable. We recommend that you include plots showing the performance change as a function of overlap criteria when appropriate. You should determine and fix the detection accuracy assessment methodology, including the selection of primary and secondary performance endpoints, prior to initiating your evaluation.
All performance measures should be reported with associated confidence intervals (CIs). We recommend that you provide a description of your methodology for estimating these CIs and the clinical significance associated with these CIs.
We also recommend that you provide graphs of the standalone free-response receiver operating characteristic (FROC) curves (i.e., a plot of patient-based standalone sensitivity vs. average number of FPs/case as a function of operating point) when reporting detection accuracy and the clinical interpretation of this analysis. You should report associated standalone FROC CIs when appropriate. Resampling techniques, such as the bootstrap,15 are potential methodologies for estimating these CIs.
Finally, we recommend that your testing accounts for differences in both the location and extent of the abnormality (e.g., ratio of the intersection area of CADe mark and reference standard to the union of these areas) when your marker includes information about both of these factors (e.g., CADe marker is outline of the segmented structure).
D. Generalizability Testing 🔗
We recommend that you justify the extent to which your studies generalize to the range of acquisition technologies defined in the device labeling. This may include studying the impact of various acquisition technologies and acquisition parameters on CADe performance to provide insight into the stability of the CADe algorithm. You should conduct this additional testing in a way that is appropriate and that does not instill additional risk (e.g., increase radiation or contrast exposure) to the patient while still examining the impact of various image acquisition confounders, such as:
- imaging hardware,
- imaging or scanning protocol, and
- image or data characteristics (e.g., characteristics associated with differences in digitization architectures for a CADe using scanned films).
Examples of generalizability testing for CADe device include performing:
- a stratified analysis of parameters expected to introduce variability in the results (e.g., scanning characteristics, make and model of the imaging devices, acquisition protocol parameters such as contrast agent or probe positioning) when the data used in the development and testing reflect the imaging requirements;
- a sound technical argument or bench-top data to support compatibility with your CADe device for various imaging systems that use the same or similar underlying technology;
- a performance comparison for a set of images digitized multiple times on different scanners when the CADe input is digitized image data;
- a performance assessment of multiple image acquisitions from the same patient with the same modalities (e.g., MRI or ultrasound). Again, any additional testing should be conducted in a way that is appropriate and that does not instill additional risk to the patient; and
- a performance assessment of simulated data in lieu of actual acquisitions (e.g., parametric noise simulation studies, simulated reconstructions, resolution reduction) when the validity of the simulated data has been established.
We recommend that you provide the following in your generalizability evaluation:
- an evaluation of parameters expected to introduce variability in the results (e.g., scanning characteristics, make and model of the imaging devices, acquisition protocol parameters, such as contrast agent or probe positioning) and their effect on CADe performance;
- a description of the studies conducted; and
- the results and statistical analysis for your evaluation.
We also recommend that you identify the specific devices or device technologies and acquisition techniques that you use in validation testing and include the specific devices or device technologies and acquisition techniques that are compatible with your CADe device in the product labeling.
E. Comparison with Predicate Device 🔗
A direct comparison of the standalone performances of your device and that of the predicate device (i.e., comparing performance using the same evaluation process and test data set) is recommended, if such a comparison is feasible. A direct comparison to a predicate device from another manufacturer may not always be possible. However, it is expected that, if you submit a 510(k) for a modified device based on your own device, you should have access to previously cleared versions of the device, and should therefore be able to perform this comparison. We recommend that you describe your comparison analysis, hypothesis to be tested, sample size estimation, and endpoints, and provide comparison results including CADe standalone performance measure such as:
- difference in area under the standalone FROC curves with associated statistical analysis (e.g., see Samuelson et al.16); or
- difference in detection standalone sensitivity and the number of FPs/case at the device operating points, and
- a comparison of the true positive and false positive CADe marks in both location (and extent when applicable) between the new or changed CADe device and the predicate (e.g., average percent of overlapping true positive marks prompted by both devices, average percent of overlapping false positive marks prompted by both devices, and an analysis of the size for the CADe marks produced by each device) when a direct comparison between devices is possible.
The goal of the latter comparison is to determine whether both the true positive and false positive CADe marks are consistent with the corresponding marks produced by the predicate device. We recommend that you evaluate this important additional factor because two CADe devices with similar standalone sensitivity and false positive rates may mark different structures within an image and this difference may result in a meaningful difference in clinical performance.
Again, the standalone data set should be large enough such that the study has adequate power to detect with statistical significance your proposed performance claims. Non-inferiority studies may be appropriate in some situations to facilitate comparisons. As mentioned above, the study should contain a sufficient number of cases from important cohorts (e.g., subsets defined by clinically relevant confounders, effect modifiers, and concomitant diseases) such that you can obtain standalone performance estimates and confidence intervals for these individual subsets (e.g., performance estimates for different nodule size categories when evaluating a lung CADe device). Powering these subsets for statistical significance should not be necessary unless you are making specific subset performance claims.
Reporting of standalone performance results may be guided by the FDA guidance entitled “Statistical Guidance on Reporting Results from Studies Evaluating Diagnostic Tests.”17
F. Electronic Data 🔗
We recommend submitting the data used in any statistical analysis in your study including patient information, disease or normal status, lesion size, lesion type, imaging and scanning setting, and imaging and data characteristics electronically (e.g., on a CD-ROM) when possible to streamline device review. For more information on submitting data electronically, please see the FDA white paper entitled Clinical Data for Premarket Submissions, and also FDA’s webpage on the eCopy Program18 and the FDA guidance entitled “eCopy Program for Medical Device Submissions.”19
VI. Clinical Performance Assessment 🔗
As described above, because each new CADe device represents a new implementation of the CADe algorithm and software, FDA expects that each new CADe device may have different technological characteristics from the legally marketed predicate device even while sharing the same intended use. Accordingly, under section 513(i)(1)(A) of the FD&C Act, determinations of substantial equivalence will rest on whether the information submitted, including appropriate clinical or scientific data, demonstrate that the new or changed device is as safe and effective as the legally marketed predicate device and does not raise different questions of safety and effectiveness than the predicate device.
There may be circumstances where a clinical performance assessment may not be necessary to demonstrate that that the new or changed device is as safe and effective as the legally marketed predicate device and does not raise different questions of safety and effectiveness from that of the predicate device. This may occur if you are able to directly compare your standalone performance to the corresponding performances of the legally marketed predicate device to which you are claiming substantial equivalence (i.e., compare standalone performance using the same evaluation process and test data set). You may not need to conduct a new clinical performance assessment study if you can make a direct comparison and you can show that the new or changed device has at least one of the following:
- an improvement in the standalone sensitivity metric (e.g., increased lesion-based sensitivity), and a stable or improved standalone false positive rate metric;
- an improvement in the standalone false positive rate metric (e.g., reduction in the number of false positives per case), and a stable or improved standalone sensitivity metric; or
- an improvement in the standalone FROC curves;
and the new or changed device produces both true positive and false positive CADe marks that are reasonably consistent with the marks produced by the predicate device in both location and extent.
We believe that the most likely scenario for when you should not need to conduct a new clinical performance study is where you are able to compare a modified CADe device to the original previously-released version of the device. The Agency believes that it could be extremely difficult to demonstrate substantial equivalence between the new or changed device and the predicate based on an indirect comparison because of potential differences (e.g., in the datasets, reference standards, scoring processes, evaluation methodologies) in the two studies.
There may be other situations where a standalone performance assessment without a clinical performance assessment (i.e., a reader study) may be sufficient to demonstrate substantial equivalence. If you believe this might apply to your device, we recommend you contact the Agency to discuss your situation.
Because the reader is an integral part of the diagnostic process for CADe devices, we believe that a standalone performance assessment without a clinical performance assessment (i.e., a reader study) may not be adequate to demonstrate that the diagnostic performance of the CADe device is as safe and effective as the legally marketed predicate when the standalone performance cannot be directly compared to that of the predicate device. In addition, a direct comparison of the overall standalone performance with the predicate device may not be adequate when there has been a change to the indications for use or when there are substantive technological differences with the predicate (e.g., resulting in marks with substantially different characteristics from that of the predicate device). In these situations, you should assume that a clinical assessment should be conducted as necessary to demonstrate substantial equivalence between your CADe device and its predicate for its intended use, when used by the intended user and in accordance with its proposed labeling and instructions.
This clinical performance assessment should provide an estimate of the clinical effect of the CADe device on clinician performance. If you believe a clinical assessment may not be necessary for demonstrating substantial equivalence of your device with the predicate, we recommend that you contact the Agency to seek advice prior to conducting your studies.
For clinical assessment, we recommend that you assess the clinical performance of your CADe device relative to a control modality. Various control arms can be employed with the most common approach being unaided reading. The use of unaided reading as the control provides an assessment of the clinical effectiveness of your device, which, in 510(k) studies, should be compared with the clinical effectiveness of the predicate device, as estimated in a prior study. Another example of a control could be the use of the predicate device evaluated on the same data set as your device. In this case you should directly compare the performance of clinicians using your CADe device with the performance of the same clinicians using the predicate device on the same data set (see Section VI.A. Comparison with Predicate Device for more details on comparing clinical performance to that of the predicate device).
For further detail on potential clinical assessment methodologies, we recommend that you consult the guidance entitled “Clinical Performance Assessment: Considerations for Computer-Assisted Detection Devices Applied to Radiology Images and Radiology Device Data in Premarket Notification (510(k)) Submissions.”20
Examples of changes to an already cleared CADe device for which we recommend submitting a clinical performance assessment include:
- the characteristics or makeup of the database used to assess standalone performance (see Section V. Standalone Performance Assessment) cannot be demonstrated to be comparable to the characteristics or makeup of the database used in assessing the predicate device and these differences raise clinical concerns (i.e., could significantly affect safety or effectiveness);
- the results of the standalone performance assessment (see Section V. Standalone Performance Assessment) are inferior to those of the predicate device;
- the location or extent of the CADe marks are substantially different from those of the predicate device, and these differences raise clinical concerns (i.e., could significantly affect safety or effectiveness);
- the reference standard definition, scoring process, analysis methodology, or performance endpoints are different from those of the predicate device, and the significance and effect on the clinician or patient of these differences are not well-known or well-described in the literature;
- the algorithm design is different from that of the predicate device and this difference raises clinical concerns (i.e., could significantly affect safety or effectiveness); and
- a new acquisition technology or protocol is employed, changing the nature of the inputs to the CADe (e.g., the current CADe device is applied to digital radiographs whereas the predicate device was applied to film-based radiographs) and these differences raise clinical concerns (i.e., could significantly affect safety or effectiveness).
We recommend that a standalone performance assessment be included in your submission regardless of whether a clinical performance assessment is included.
A. Comparison with Predicate Device 🔗
We recommend that you compare your clinical performance assessment results to those of the predicate device to which you are claiming substantial equivalence (e.g., a previously released version of the device) when a clinical assessment is necessary. A direct comparison to a predicate device may not always be possible. In this case, an alternative study could be a comparison to a control arm that establishes that the use of your device for its intended use, when used by the intended user and in accordance with its proposed labeling and instructions, leads to a statistically significant improvement in performance over that in the control arm (e.g., with CADe reading is statistically superior to without CADe reading). The performance results would then be compared to the corresponding results for a predicate that underwent a similar clinical assessment and showed a statistically significant improvement in performance over that of the control. This type of alternative clinical performance assessment study can be used when a direct comparison of clinical performance between your device and the predicate is not possible.
Reporting of clinical performance results may be guided by the FDA guidance entitled “Statistical Guidance on Reporting Results from Studies Evaluating Diagnostic Tests.”21
B. Electronic Data 🔗
We recommend submitting the data used in any statistical analysis in your clinical assessment study including patient information, disease or normal status, lesion size, lesion type, imaging and scanning setting, and imaging and data characteristics electronically (e.g., on a CD-ROM) when possible to streamline device review. For more information on submitting data electronically, please see the FDA webpage on the eCopy Program22 and the FDA guidance entitled “eCopy Program for Medical Device Submissions.”23
VII. User Training 🔗
We recommend that you provide a summary of the procedure that will be used to train the intended users of your device when marketed. The goal of this training should be to help clinicians use the CADe device in an appropriate manner and to provide training so that they can achieve the expected device effectiveness. Training should include both the expected advantages and known limitations of the device (e.g., the CADe does not identify calcified nodules). Training should be based on a broad set of patient data including normal cases. This training data should include typical true positives and false positive that the device tends to output, as well as typical true negatives and false negatives.
For CADe devices allowing multiple thresholds or operating points, the training should help clinicians identify the most appropriate device setting for their practices. In addition, the training should help clinicians identify suitable CADe reading scenarios.
VIII. Labeling 🔗
In accordance with 21 CFR 807.87(e), you must provide proposed labels, labeling, and advertisements sufficient to describe the device, the intended use, and the directions for its use. The 510(k) must include labeling in sufficient detail to satisfy the requirements of 21 CFR 807.87(e). The following suggestions are aimed at assisting you in preparing labeling that satisfies the requirements of 21 CFR Part 801.19.24
Your user manual should include the information described below.
A. Indications For Use 🔗
We recommend that the indications for use (IFU) address how the device will be used, for example:
The device is intended to assist [target users] in their review of [patient/data characteristics] in the detection of [target disease/condition/abnormality] using [image type/technique and conditions of imaging].
B. Directions For Use 🔗
There must be adequate directions for use as described in 21 CFR 801.5; the requirements applicable to prescription devices are described in 21 CFR 801.109. You should submit clear and concise instructions that delineate the technological features of the specific device and how the device is to be used on patient images/data. Instructions should encourage local/institutional training programs designed to familiarize clinicians with the features of the device and how to use it in a safe and effective manner. The directions should also clearly define the intended user of the device.
C. Warnings 🔗
The warnings should address limitations of the device. For example:
[Target user] should not rely solely on the output identified by [**device trade name], but should perform a full systematic review and interpretation of the entire patient dataset.
Another example may be:
This CADe device has been found to be ineffective for patients with [disease/condition/ abnormality]. This CADe should not be utilized with patients presenting with this [disease/condition/abnormality].
D. Precautions 🔗
The precautions should discuss the potential for adverse events associated with the use of the device and recommend mitigation measures. The adverse event discussion should at least include a discussion of potential adverse events associated with an increased workup rate (i.e., events from false-positives) and missed disease/condition/abnormality.
E. Device Description 🔗
We recommend that you include the following in your device description:
- an overview of the algorithm design and features;
- an overview of the training paradigm and the training or development database;
- a description of the reference standard used for patient data utilized in the development and adjustment of the algorithm;
- specific devices or device technologies and acquisition techniques compatible with your CADe device; and
- the display requirements for appropriately displaying your CADe marks for devices designed as add-on software for general image review workstations and PACS.
F. Clinical Performance Assessment 🔗
When appropriate, we recommend that you include a summary of the clinical performance assessment including:
- study objectives,
- study design,
- patient population (e.g., age, ethnicity, race, sex),
- number of clinicians and their qualification,
- description of the methodology used in gathering clinical information,
- description of the statistical methods used to analyze the data, and
- study results.
Additional information on reporting clinical performance results can be found in the FDA guidance entitled “Clinical Performance Assessment: Considerations for Computer-Assisted Detection Devices Applied to Radiology Images and Radiology Device Data in Premarket Notification (510(k)) Submissions.”24
G. Standalone Performance Assessment 🔗
We recommend that you provide a summary of the device standalone performance and generalizability testing including:
- the scoring criteria used to determine the nature of each region marked by your CADe device;
- the overall standalone sensitivity and standalone false positive rate metrics at each available device operating point;
- stratified analysis (e.g., per lesion size, per lesion type, per imaging or scanning protocols, per imaging or data characteristics), as appropriate;
- the CIs on each measure; and
- the standalone FROC performance, as appropriate.
Footnote 🔗
-
The use of the acronym CADe for computer-assisted detection may not be a generally recognized acronym in the community at large. It is used here to identify the specific type of devices discussed in this document. ↩
-
Available at https://wayback.archive-it.org/7993/20170405191951/https:/www.fda.gov/AdvisoryCommittees/CommitteesMeetingMaterials/MedicalDevi ces/MedicalDevicesAdvisoryCommittee/RadiologicalDevicesPanel/ucm124890.htm. ↩
-
Available at https://wayback.archive-it.org/7993/20170405191949/https:/www.fda.gov/AdvisoryCommittees/CommitteesMeetingMaterials/MedicalDevices/MedicalDevicesAdvisoryCommittee/RadiologicalDevicesPanel/ucm147063.htm. ↩
-
See 74 FR 54053 at https://www.federalregister.gov/documents/2009/10/21/E9-25233/draft-guidances-for-industry-and-food-and-drug-administration-staff-computer-assisted-detection. ↩
-
Available at https://www.fda.gov/regulatory-information/search-fda-guidance-documents/format-traditional-and-abbreviated-510ks. ↩
-
See 86 FR 20278 at https://www.federalregister.gov/documents/2021/04/19/2021-07860/medical-devices-medical-device-classification-regulations-to-conform-to-medical-software-provisions. ↩
-
For more information on product codes, please refer to FDA’s guidance entitled “Medical Device Classification Product Codes” available at https://www.fda.gov/regulatory-information/search-fda-guidance-documents/medical-device-classification-product-codes-guidance-industry-and-food-and-drug-administration-staff. ↩
-
Available at https://www.fda.gov/regulatory-information/search-fda-guidance-documents/clinical-performance-assessment-considerations-computer-assisted-detection-devices-applied-radiology. ↩
-
Available at https://www.fda.gov/regulatory-information/search-fda-guidance-documents/guidance-content-premarket-submissions-software-contained-medical-devices. ↩
-
Available at https://www.fda.gov/regulatory-information/search-fda-guidance-documents/shelf-software-use-medical-devices. ↩
-
21 CFR 807.81(a)(3). ↩
-
Available at https://www.fda.gov/regulatory-information/search-fda-guidance-documents/establishment-and-operation-clinical-trial-data-monitoring-committees. ↩
-
Miller, D.P., O’Shaughnessy, K.F., Wood, S.A., and Castellino M.D., R.A., Gold standards and expert panels: a pulmonary nodule case study with challenges and solutions. Proc. SPIE 5372, Medical Imaging 2004: Image Perception, Observer Performance, and Technology Assessment. https://doi.org/10.1117/12.544716. ↩
-
Standalone specificity (i.e., the fraction of normal cases without a CADe mark) may be applicable instead of standalone false positive rate for some CADe algorithms that have very few false positive detections. ↩
-
Efron, B.,Tibshirani, R., Bootstrap Methods for Standard Errors, Confidence Intervals, and Other Measures of Statistical Accuracy. Statist. Sci., 1986. 1(1) 54-75. https://doi.org/10.1214/ss/1177013815. ↩
-
Samuelson, F.W., and Petrick, N., Comparing image detection algorithms using resampling. 3rd IEEE International Symposium on Biomedical Imaging: Nano to Macro, 2006. 2006:1312-1315. https://doi.org/10.1109/ISBI.2006.1625167. ↩
-
Available at https://www.fda.gov/regulatory-information/search-fda-guidance-documents/statistical-guidance-reporting-results-studies-evaluating-diagnostic-tests-guidance-industry-and-fda. ↩
-
Available at https://www.fda.gov/medical-devices/how-study-and-market-your-device/ecopy-program-medical-device-submissions. ↩
-
Available at https://www.fda.gov/regulatory-information/search-fda-guidance-documents/ecopy-program-medical-device-submissions. ↩
-
Available at https://www.fda.gov/regulatory-information/search-fda-guidance-documents/clinical-performance-assessment-considerations-computer-assisted-detection-devices-applied-radiology. ↩
-
Available at https://www.fda.gov/regulatory-information/search-fda-guidance-documents/statistical-guidance-reporting-results-studies-evaluating-diagnostic-tests-guidance-industry-and-fda. ↩
-
Available at https://www.fda.gov/medical-devices/how-study-and-market-your-device/ecopy-program-medical-device-submissions. ↩
-
Available at https://www.fda.gov/regulatory-information/search-fda-guidance-documents/ecopy-program-medical-device-submissions. ↩
-
Available at https://www.fda.gov/regulatory-information/search-fda-guidance-documents/clinical-performance-assessment-considerations-computer-assisted-detection-devices-applied-radiology. ↩
