
Innolitics Introduction 🔗
Software is, well, “softer” than hardware. It can be changed more easily. It also needs to change more often in response to changes in the interoperability or cybersecurity environment.
It is therefore more common that medical device software changes, and manufacturers must decide for each change, whether they need to submit a new 510(k).
Understanding this guidance is essential for planning your software development road map in a way that you can reduce the regulatory burden of your project. In particular, you want to lump changes that will trigger a 510(k) together.
If you’re looking for a regulatory consulting firm that understands software, we can help. We can help refine your regulatory and software development strategy to minimize your costs.
About this Transcript 🔗
This document is a transcript of an official FDA (or IMDRF) guidance document. We transcribe the official PDFs into HTML so that we can share links to particular sections of the guidance when communicating internally and with our clients. We do our best to be accurate and have a thorough review process, but occasionally mistakes slip through. If you notice a typo, please email a screenshot of it to Mihajlo at mgrcic@innolitics.com so we can fix it.
Preamble 🔗
Document issued on October 25, 2017.
The draft of this document was issued on August 8, 2016.
For questions about this document, contact (CDRH) Linda Ricci, Office of Device Evaluation, 301-796-6325, Linda.Ricci@fda.hhs.gov.
For questions about this document regarding CBER-regulated devices, contact the Office of Communication, Outreach and Development (OCOD), by calling 1-800-835-4709 or 240-402-8010.
Contains non-binding guidance.
I. Introduction 🔗
This guidance will assist industry and Agency staff in determining when a software (including firmware) change to a medical device may require a manufacturer to submit and obtain FDA clearance of a new premarket notification (510(k)). This guidance is not intended to implement significant policy changes to FDA’s current thinking on when submission of a new 510(k) is required for a software change to a 510(k)-cleared device (or group of devices) or other device subject to 510(k) requirements, such as a preamendments device or a device that was granted marketing authorization via the De Novo classification process under section 513(f)(2) of the Food, Drug, and Cosmetic Act (FD&C Act) (also referred to together as “existing devices”). Rather, the intent of this guidance is to enhance the predictability, consistency, and transparency of the “when to submit” decision-making process by providing a least burdensome approach, and describing in greater detail the regulatory framework, policies, and practices underlying such a decision, specifically as it relates to software changes.
For the current edition of the FDA-recognized standards referenced in this document, see the FDA Recognized Consensus Standards Database at http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfStandards/search.cfm.
FDA's guidance documents, including this guidance, do not establish legally enforceable responsibilities. Instead, guidances describe the Agency’s current thinking on a topic and should be viewed only as recommendations, unless specific regulatory or statutory requirements are cited. The use of the word should in Agency guidance means that something is suggested or recommended, but not required.
II. Background 🔗
The regulatory criteria in 21 CFR 807.81(a)(3) state that a premarket notification must be submitted when:
(3)The device is one that the person currently has in commercial distribution or is reintroducing into commercial distribution, but that is about to be significantly changed or modified in design, components, method of manufacture, or intended use. The following constitute significant changes or modifications that require a premarket notification:
- A change or modification in the device that could significantly affect the safety or effectiveness of the device, e.g., a significant change or modification in design, material, chemical composition, energy source, or manufacturing process.
- A major change or modification in the intended use of the device.
FDA issued the original guidance Deciding When to Submit a 510(k) for a Change to an Existing Device (K97-1) on January 10, 1997 to provide guidance on this regulatory language. As stated in that guidance, the key issue in the interpretation of 21 CFR 807.81(a)(3) is that the phrase “could significantly affect the safety or effectiveness of the device” and the use of the adjectives "major" and "significant" sometimes lead FDA and device manufacturers to different interpretations. The original guidance provided the Agency’s interpretation of these terms, with principles and points for manufacturers to consider in analyzing how changes in devices may affect safety or effectiveness and determining whether a new 510(k) must be submitted for a particular type of change. The current guidance preserves the basic format and content of the original, with updates to add clarity. The added clarity is intended to increase consistent interpretations of the guidance by FDA staff and manufacturers and provide a more transparent framework for determining when submission of a new 510(k) is required.
The 510(k) Process and the Quality System Regulation 🔗
Any guidance on 510(k)s for changes to a legally marketed device should consider the role the Quality System (QS) regulation, 21 CFR Part 820, plays in changes to devices. For some types of changes to a device, the Agency believes that submission of a new 510(k) is not required and that reliance on existing QS requirements is the least burdensome approach to reasonably assure the safety and effectiveness of the changed device.
Regardless of whether a change requires premarket review, the QS regulation requires manufacturers of finished medical devices to review and approve changes to device design and production (21 CFR 820.30 and 820.70) and document changes and approvals in the device master record (21 CFR 820.181). Any process whose results cannot be fully verified by subsequent inspection and testing must be validated (21 CFR 820.75), and changes to the process require review, evaluation, and revalidation of the process where appropriate (21 CFR 820.75(c)).
The net effect of the QS regulation is to require that, when manufacturers of a finished medical device make a change in the design of a device, there is a process in place to demonstrate that the manufactured device meets the change in design specifications (or the original specifications, if no change was intended). They must keep records, and these records must be made available to an FDA investigator upon request (see Section 704(e) of the FD&C Act). For many changes to a device, submission of a new 510(k) may not be required. In these cases, including many software design changes, compliance with the QS regulation can reasonably assure the safety and effectiveness of the changed device.
Least Burdensome Principles 🔗
The least burdensome provision concerning 510(k)s states that FDA “shall only request information that is necessary…” and “shall consider the least burdensome means of demonstrating substantial equivalence…” (see section 513(i)(1)(D)(i) of the FD&C Act). While not changing the standard for substantial equivalence, this provision states that FDA shall only request the “minimum required information” necessary to support a determination of substantial equivalence (see sections 513(i)(1)(D)(ii)-(iii) of the FD&C Act). The recommendations discussed in this guidance for evaluating when a change in a medical device would trigger the requirement that a manufacturer submit a new 510(k) to the Agency are consistent with least burdensome principles, and applies them in discussing the considerations that may affect the decision-making about when to submit a new 510(k) for a device change or modification.
III. Scope 🔗
As used in this guidance, “software” is the set of electronic instructions used to control the actions or output of a medical device, to provide input to or output from a medical device, or to provide the actions of a medical device. This definition includes software that is embedded within or a component of a medical device, software that is an accessory to another medical device, or software that is intended to be used for one or more medical purposes that performs these purposes without being part of a hardware medical device.1
This guidance will aid manufacturers of medical devices subject to premarket notification requirements who intend to modify a 510(k)-cleared device (or group of devices) or other device subject to 510(k) requirements, such as a preamendments device or a device that was granted marketing authorization via the De Novo classification process2 under section 513(f)(2) of the FD&C Act (also referred to together as “existing devices”), during the process of deciding whether the change exceeds the regulatory threshold of 21 CFR 807.81(a)(3) for submission and clearance of a new 510(k). Note that any person required to register under 21 CFR 807.20 who plans to introduce a device into commercial distribution for the first time must, per 21 CFR 807.81(a)(2), submit a 510(k) if that device is not exempt from premarket notification requirements. Also note that devices with changes requiring submission of a new 510(k) may not be legally commercially distributed before FDA clears the changed device (21 CFR 807.100(a) and sections 513(f)(1) and 513(i) of the FD&C Act). Private label distributors and repackagers are exempt from submitting a 510(k) if they satisfy the requirements of 21 CFR 807.85(b). This guidance is not intended to address changes to devices that are 510(k)-exempt or that require premarket approval (PMA).
This guidance specifically addresses software modifications and is intended as a companion to Deciding When to Submit a 510(k) for a Change to an Existing Device (http://www.fda.gov/downloads/MedicalDevices/DeviceRegulationandGuidance/GuidanceDocuments/ucm080243.pdf). Any modifications that are not modifications to software are not within the scope of this guidance; such changes (e.g., labeling changes) should be evaluated using Deciding When to Submit a 510(k) for a Change to an Existing Device. Both guidance documents explain FDA’s current thinking on 21 CFR 807.81(a)(3), and as such, the threshold for submission of a new 510(k) in response to a change to an existing device is not different between the two guidances; however the terminology used may differ due to the nature of the technology and the assessment of the risks associated with the change. In addition, it may be necessary to refer to other relevant FDA guidance documents that aid in the evaluation of non- software device modifications. It is the manufacturer’s responsibility to collectively evaluate the combination of both software and non-software changes to evaluate the impact of a change to a device. For those circumstances where the proposed change is not addressed in this guidance, in Deciding When to Submit a 510(k) for a Change to an Existing Device, or in a device-specific guidance, manufacturers are encouraged to contact the appropriate office in CDRH or CBER.
When there are multiple changes that affect labeling or hardware in addition to software, the manufacturer should assess the changes using both the general and software-specific modifications guidances. If use of either guidance leads to a “New 510(k)” conclusion, submission of a new 510(k) is likely required.
This guidance does not apply to software for which the Agency has stated in guidance that it does not intend to enforce compliance with applicable regulatory controls (see, e.g., Mobile Medical Applications Guidance for Industry and FDA Staff https://www.fda.gov/regulatory-information/search-fda-guidance-documents/policy-device-software-functions-and-mobile-medical-applications. Further, this guidance does not address the software lifecycle (covered in AAMI/ANSI/IEC 62304: Medical device software - software life cycle processes), what documentation should be included in a 510(k) for a software modification (covered in Guidance for the Content of Premarket Submissions for Software Contained in Medical Devices (https://www.fda.gov/regulatory-information/search-fda-guidance-documents/content-premarket-submissions-device-software-functions)) or the principles that are applicable to the validation of medical-device software (covered in General Principles of Software Validation; Final Guidance for Industry and FDA Staff (https://www.fda.gov/regulatory-information/search-fda-guidance-documents/general-principles-software-validation)).
This guidance is also intended to apply to situations when a legally marketed existing device is the subject of a recall, correction, or removal, and a change in the device or its labeling is necessary. For more information on recommended procedures in a recall situation, please see Blue Book Memorandum K95-1, 510(k) Requirements During Firm-Initiated Recalls (http://www.fda.gov/MedicalDevices/DeviceRegulationandGuidance/GuidanceDocuments/ucm080297.htm). As stated in that guidance, if a correction alters a device rather than simply restoring it to its original specifications, submission of a new 510(k) may be required. FDA may use this guidance in determining whether submission of a new 510(k) is warranted in cases where the correction does alter the device.
This guidance does not specifically address combination products, such as drug/device or biologic/device combinations; however, the general principles and concepts described herein may be helpful to manufacturers in determining whether submission of a 510(k) is required for changes to software-containing device constituent parts of combination products.
Software modifications may be identified by many names, including, but not limited to: bug fix, hot fix, patch, software change, code change, or tweak. Regardless of name or form, these are considered design changes under the Quality System regulation, 21 CFR Part 820.
This guidance is not intended to supersede final device-specific guidance (such as the Infusion Pumps Total Product Life Cycle https://www.fda.gov/regulatory-information/search-fda-guidance-documents/infusion-pumps-total-product-life-cycle), but may cover areas not addressed in any final device-specific guidance.
IV. Guiding Principles 🔗
In using this guidance for deciding whether to submit a new 510(k) for a change to an existing device, a number of guiding principles should be followed. Some derive from existing FDA 510(k) policy and are widely known, and others are necessary for using the logic scheme contained in this guidance. Thus, anyone using this guidance should bear in mind the following Guiding Principles:
-
Changes made with intent to significantly affect safety or effectiveness of a device – If a manufacturer modifies their device with the intent to significantly affect the safety or effectiveness of the device (for example, to significantly improve clinical outcomes, to mitigate a known risk, in response to adverse events, etc.), submission of a new 510(k) is likely required. A change intended to significantly affect the safety or effectiveness of the device is considered to be a change that “could significantly affect the safety or effectiveness of the device” and thus requires submission of a new 510(k) regardless of the considerations outlined below. Changes that are not intended to significantly affect the safety or effectiveness of a device, however, should still be evaluated to determine whether the change could significantly affect device safety or effectiveness. If a manufacturer modifies their device to address a violation or recall, they should refer to FDA guidances Blue Book Memorandum K95-1, 510(k) Requirements During Firm- Initiated Recalls
(http://www.fda.gov/MedicalDevices/DeviceRegulationandGuidance/GuidanceDocuments/ucm080297.htm) and Distinguishing Medical Device Recalls from Medical Device Enhancements (https://www.fda.gov/downloads/medicaldevices/deviceregulationandguidance/guidancedocuments/ucm418469.pdf).
- Initial risk-based assessment – To determine whether a change or modification could significantly affect the safety or effectiveness of a device, the manufacturer should first conduct a risk-based assessment, using the guidance below, of whether the change could significantly affect the device’s safety or effectiveness, either positively or negatively. This risk-based assessment should identify and analyze all new risks and changes in existing risks resulting from the device change, and lead to an initial decision whether or not submission of a new 510(k) is required. For the purposes of this guidance, we have chosen the term “risk-based assessment” to describe the analysis that should be completed to assist in the determination of whether or not a change could significantly affect safety or effectiveness of the device. Although common risk analysis methods define risk in terms of device harms and their effects on safety, it is important to note that whether submission of a new 510(k) is required depends on whether the change could significantly affect the safety or effectiveness of the device. Therefore, manufacturers should also consider the possible effects a device change may have on device effectiveness. As such, we have chosen to use the distinct terminology of “risk-based assessment.”
- Unintended consequences of changes – After a manufacturer considers whether the change was made with the intent to significantly affect safety or effectiveness, the manufacturer should also consider whether the change could have unintended consequences. Software modifications may trigger additional unintended or unplanned consequences which should be assessed using the flowchart (and its companion text) to determine if submission of a new 510(k) is needed. For example, an intended operating system (OS) upgrade may trigger unintended effects in device drivers and software code embedded in the device and/or may require an update to other components for compatibility purposes. Manufacturers should consider all consequences of changes to fully assess whether submission of a new 510(k) is required.
- Use of risk management – A risk-based assessment as referred to throughout this document is based on the combination of multiple risk concepts that are important for managing the risks of medical devices. Hazards and hazardous situations, risk estimation, risk acceptability, risk control, risk/benefit analysis and overall risk evaluation are all concepts that can be applied during the design and development of a medical device. The concept of risk, as defined in ISO 14971: Medical devices – Application of risk management to medical devices, is the combination of the probability of occurrence of harm and the severity of that harm. Although the risk terminology used in this document is primarily derived from ISO 14971, we recognize that an individual manufacturer’s terminology may differ. Because 21 CFR 807.81(a)(3)(i) requires submission of a new 510(k) when a change “could significantly affect safety or effectiveness,” both safety and effectiveness should be considered in evaluating a device’s risk profile and performing a risk-based assessment. The risk terminology from the currently FDA-recognized version of IEC TR 80002-1: Medical device software – Part 1: Guidance on the application of ISO 14971 to medical device software is also used in this guidance. For software, failures tend to be systematic in nature and therefore the probability of occurrence of a software failure cannot be determined using traditional statistical methods. While it may be possible to estimate the probability for other events in the sequence, if the overall probability of occurrence of harm cannot be estimated, the estimation of risk should be based on the severity of harm alone.
- The role of testing (i.e., verification and validation activities) in evaluating whether a change could significantly affect safety and effectiveness – If the initial decision following the risk-based assessment is that submission of a new 510(k) is not required, this decision should be confirmed by successful, routine verification and validation activities. If routine verification and validation activities produce any unexpected results, any prior decision that submission of a new 510(k) is not required should be reconsidered in light of these issues (i.e., go through the flowchart again). Because 21 CFR 807.81(a)(3) requires submission of a new 510(k) for a change that “could significantly affect safety or effectiveness,” if the result of a risk-based assessment is that a change could significantly affect safety or effectiveness, submission of a new 510(k) is required even if routine verification and validation activities are conducted successfully without any unexpected results. Note that verification and validation requirements apply for all devices subject to 21 CFR 820.30, and must be conducted regardless of whether submission of a new 510(k) is required.
- Evaluating simultaneous changes to determine whether submission of a new 510(k) is required – Because many simultaneous changes may be considered at once, each change should be assessed separately, as well as in aggregate. Note that, for software, each individual line change in the code may not constitute an individual change in the device.
- Appropriate comparative device and cumulative effect of changes – In using this guidance to help determine whether a particular change requires submission of a new 510(k), a manufacturer should conduct a risk-based assessment that compares the changed device to their device as previously found to be substantially equivalent in their most recently cleared 510(k), to their preamendments device (if the device was in commercial distribution before May 28, 1976 and there have not been changes to it subsequently cleared in a 510(k)), or to their device that FDA granted marketing authorization via the De Novo classification process (if there have not been changes to it subsequently cleared in a 510(k)). The appropriate comparative device is referred to as the “original device” throughout this guidance document. Of note, this comparison is different from a substantial equivalence comparison between the modified device and a legally marketed predicate device. Manufacturers may make a number of changes without having to submit a new 510(k), but each time they make a change, the modified device should be compared to the original device (i.e., the device described in their most recently cleared 510(k) for the device, to their legally marketed preamendments device, or to their device that was granted marketing authorization via the De Novo classification process). When the cumulative effect of individual changes triggers the regulatory threshold for submission, the manufacturer should submit a new 510(k). When it does not, the manufacturer must document the change(s) (see 21 CFR Part 820.30).
- Documentation requirement – Whenever manufacturers change their device, they must take certain actions to comply with the QS regulation, 21 CFR Part 820, unless the device in question is exempt by regulation from the QS regulation. The QS regulation requires, among other things, that device changes be documented. The scope and type of documentation may vary, but the process of documenting the decisions described in this guidance should be established as part of the manufacturer’s own quality system.
- 510(k) submissions for modified devices – When a new 510(k) is submitted for a device with multiple changes, that 510(k) should describe all changes that trigger the requirement for submission of a new 510(k). To help ensure that FDA has a complete understanding of the device under review, that 510(k) should also describe other changes since the most recently cleared 510(k) (i.e., those that did not require submission of a new 510(k)) that would have been documented as part of the first 510(k) for that device. For instance, 510(k)s typically include a listing of device warnings in the labeling, so if a warning in the device’s labeling had been changed, that change should be described in the new 510(k) for the software modification even if that labeling change did not itself trigger the requirement for submission of a new 510(k) and the 510(k) is being triggered by a software modification only. A 510(k) would not typically identify or describe individual components of a circuit board, such as resistors, or identify the specific version of a printer driver, and therefore FDA would not expect changes to the resistors or the printer driver to be listed in the new 510(k) for a modified device because the first 510(k) would have not included information about the resistors or printer drivers. Please note that manufacturers should know which versions of off-the-shelf software and/or firmware are included in their device even if that level of detail is not included in a 510(k). If a manufacturer makes multiple changes to a device, but only one change triggers the requirement for submission of a new 510(k), the changes that do not require submission of a new 510(k) may be immediately implemented, so long as those changes can be implemented independently of changes that do require submission of a new 510(k). Any immediately implemented change should still be documented in accordance with applicable QS regulations and the manufacturer’s documentation procedures. Those changes should, however, also be described in the new 510(k) for the change that does require submission.
- Substantial equivalence determinations – Manufacturers should understand that, even though they may follow this guidance and submit a new 510(k), a substantially equivalent determination is not assured. See FDA’s guidance The 510(k) Program: Evaluating Substantial Equivalence in Premarket Notifications (510(k)) (https://www.fda.gov/downloads/medicaldevices/deviceregulationandguidance/guidance ocuments/ucm284443.pdf) for more information on the decision-making process FDA uses to determine substantial equivalence.
V. How to Use This Guidance 🔗
This guidance uses a flowchart and text with considerations and examples to guide manufacturers through the logic scheme we recommend to arrive at a decision on whether to submit a new 510(k) for a software change to an existing device. A single logic scheme covering all the intricacies in software modifications and their impact on the decision to submit a new 510(k) would be impractical to develop. Rather, for ease of use, a flowchart and text expected to cover the most common software modifications has been created.
Manufacturers should use the flowchart in concert with the Guiding Principles above, the text below, and additional factors in section VI. Manufacturers should answer the questions posed for each individual type of change (e.g., performance specification change, OS driver change) until a decision is made either to submit a new 510(k) or to document the basis for concluding that submission of a new 510(k) is not required. As mentioned above, when making the decision on whether to submit a new 510(k) for changes, the manufacturer’s basis for comparison of any changed device should be the original device. Manufacturers are required to submit a new 510(k) when a change (or changes) exceeds the 21 CFR 807.81(a)(3) threshold, “could significantly affect the safety or effectiveness of the device,” or constitutes a “major change or modification in the intended use of the device.” This significant effect could be positive or negative. One must keep in mind that what may on the surface appear to be one discrete change to a device may involve multiple changes of various types. Appendix A provides a number of examples with rationale that can be helpful in working through this guidance.
In cases with multiple changes, manufacturers should use all applicable parts of the flowchart and companion text, including the Guiding Principles in Section IV of this guidance.
Note that the flowchart entries, “new 510(k)” and “document,” are written in this way only for conciseness. The reader should interpret “new 510(k)” as submission of a new 510(k) is likely required and “document” as a new 510(k) is likely not required, document your analysis, and file it for future reference. The goal of the flowchart is to provide guidance in answering a manufacturer’s questions on whether submission of a new 510(k) is likely required for a software change and to minimize the number of instances where the answer would be uncertain.
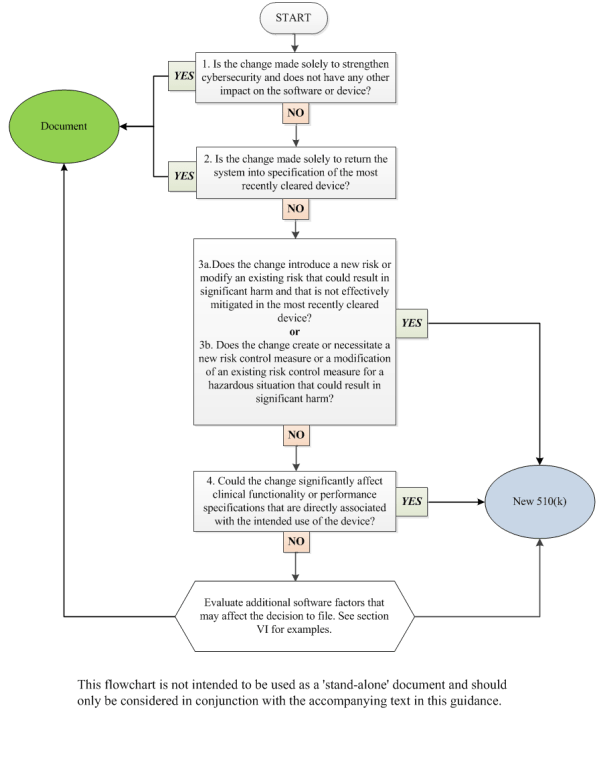
1. Is the change made solely to strengthen cybersecurity and does not have any other impact on the software or device?
In many cases, a change made solely to strengthen cybersecurity is not likely to require submission of a new 510(k). Cybersecurity updates are considered a subset of software changes that are implemented to strengthen the security of a system, protect information, and reduce disruption in service. FDA expects manufacturers to ensure that such changes do not impact the safety or effectiveness of the device by performing necessary analysis, verification, and/or validation. If a manufacturer becomes aware of any incidental or unintended impacts of the change on other aspects of the software or device, the manufacturer should continue through the remaining questions in this guidance. The manufacturer should also refer to FDA’s guidance Content of Premarket Submissions for Management of Cybersecurity in Medical Devices (http://www.fda.gov/downloads/medicaldevices/deviceregulationandguidance/guidancedocuments/ucm356190.pdf) and Postmarket Management of Cybersecurity in Medical Devices (https://www.fda.gov/downloads/medicaldevices/deviceregulationandguidance/guidancedocuments/ucm482022.pdf).
2. Is the change made solely to return the system into specification of the most recently cleared device?
When a change to the software only restores the device to the specifications of the most recently cleared device, then submission of a new 510(k) is likely not required. Generally, it is unlikely that modifications to software solely to restore the device to the most recently cleared device’s specifications could significantly impact safety, effectiveness, or intended use of the device; however, manufacturers should evaluate the impact of the software changes. Manufacturers should conduct an analysis that involves determining the overall impact of the change to the device in terms of risk assessment and performance. The concepts expressed in Questions 3 and 4 below could be helpful in this analysis. In addition, this analysis is important for evaluating any modification that adds new features that appeared in the specification of the most recently cleared device but were not yet implemented.
Missing, incomplete, ambiguous, or conflicting software requirements may lead to a software modification that involves updating specifications, resulting in additional software code changes. In these situations, the answer to this question is likely “no” and the manufacturer should proceed to Question 3.
Generally, manufacturers are not required to submit a new 510(k) for changes to a specification document to clarify to an existing software requirement or to capture a missing software requirement, provided that this does not necessitate any changes to software code or product performance specifications. However, manufacturers should still assess the impact of the changes on other software documentation when applying appropriate design controls.
3. What are the impacts of any changes to risks associated with use of the device and the impacts of any changes to the risk controls for the device?
-
Does the change introduce a new risk or modify an existing risk that could result in significant harm and that is not effectively mitigated in the most recently cleared device? The purpose of this question is to determine whether a new risk is created or has been identified, or if an existing risk is modified, as a result of the software change. The term “risk” is meant to broadly include hazard, hazardous situation, or cause of an existing hazard or hazardous situation. A “hazardous situation” exists when there is exposure to a hazard (i.e., a potential source of harm) that can lead to physical injury or damage to the health of people. The term “cause” refers to one possible component in the “sequence of events,” that can lead to a hazardous situation and possible harm, as described in ISO 14971. These are identified and defined by the manufacturer in the risk management file for the device. Significant harm refers to situations where the risk level is serious or more severe, e.g., the risk could result in injury or impairment requiring professional medical intervention, permanent impairment, or death.
Submission of a new 510(k) is likely required if all of the following criteria are met:
- The change creates a new or modifies a hazard, hazardous situation, or cause in the risk management file.
- The level of harm associated with the new or modified hazard, hazardous situation, or cause is considered serious or more severe, e.g., the hazard, hazardous situation, or cause of the hazardous situation could result in injury or impairment requiring professional medical intervention, permanent impairment, or death. The pre-mitigation risk score should be assessed in order to focus on the effects of the change.
- The hazard, hazardous situation, or cause is not already effectively mitigated in the most recently cleared device.
- Note: This criterion is met if there are no existing risk control measures in the most recently cleared device that reduce the risk associated with this hazard, hazardous situation, or cause to an acceptable level.
- Note: New hazards, hazardous situations, or causes of hazardous situations may be effectively mitigated by risk controls that were already included in the device for other hazards, hazardous situations, or causes.
If all of the above criteria are not met, proceed to Question 3b.
-
Does the change create or necessitate a new risk control measure or a modification of an existing risk control measure for a hazardous situation that could result in significant harm? It is possible that introducing new risk control measures or implementing changes to existing risk control measures could significantly affect the safety or effectiveness of the product, and thus such changes should be evaluated. It may be that the change is directly tied to the risk control measures or the software change may necessitate a new or modified risk control measure. Changes to or additions of risk control measures may be necessary due to new, modified, or previously unknown hazardous situations or causes thereof. If the changes to risk controls are necessary to prevent significant harm, submission of a new 510(k) is likely required. Conversely, submission of a new 510(k) is likely not required when implementing redundant risk control measures or enhancing existing risk control measures if the risk control measures in the most recently cleared device effectively mitigated the hazardous situation.
If the answer to this question is no, proceed to Question 4.
4. Could the change significantly affect clinical functionality or performance specifications that are directly associated with the intended use of the device?
Changes in performance specifications encompass everything from routine specification changes necessary to improve device performance to significant product redesigns. For the purpose of this question, specifications include elements that could influence the device’s ability to clinically perform as intended. These specifications may address attributes such as speed, strength, response times, throughput, limits of operation, reliability, delivery rate, or assay performance.
If the software change could significantly affect clinical functionality or performance specifications that are directly associated with the intended use of the device, then submission of a new 510(k) is likely required. For in vitro diagnostic devices (IVDs), this includes a change that could have clinically significant impact in terms of clinical decision-making. This question does not address direct changes to the indications for use and/or intended use of the device. If there is a change in the indications for use and/or intended use of the device, refer to FDA’s guidance, Deciding When to Submit a 510(k) for a Change to an Existing Device (http://www.fda.gov/downloads/MedicalDevices/DeviceRegulationandGuidance/GuidanceDocuments/ucm080243.pdf).
For IVDs, performance generally refers to the analytical and clinical specifications established as part of the most recent 510(k) clearance. Analytical performance refers to the documented ability of an IVD test or test system to measure or detect a target analyte or substance that the IVD test or test system is represented or purported to identify or measure. Clinical performance refers to the documented ability of an IVD test or test system to identify, measure, monitor, or predict the presence or absence of, or the future development of, a clinical condition or predisposition, for which the device is intended.
Depending on the assay, analytical performance specifications may be defined by:
- Analytical Sensitivity: limit of detection, reactivity(inclusivity);
- Analytical Specificity: exclusivity, cross-reactivity, interference;
- Cut-off and equivocal zone; and/or
- Precision: site-to-site reproducibility, within-laboratory precision/repeatability.
There are also times when IVD functionality or performance specifications could be changed but the change is not related to the IVD’s intended use and the performance of the modified device could not be significantly affected when compared to previously cleared performance claims, and thus submission of a new 510(k) would not be required.
VI. Additional Factors to Consider When Determining When to Submit a New 510(k) for a Software Change to an Existing Device 🔗
In addition to the questions above, the common issues below should also be considered when determining if submission of a new 510(k) is required.
Medical device software is used in a wide variety of applications and is subject to a wide variety of changes. This guidance, therefore, cannot address every type of software change. Nonetheless, the questions in the flowchart and the associated recommendations in the text provide a guide for manufacturers’ decision-making and associated documentation. The goal of the guidance is to provide examples of software changes that clearly could have a significant impact on the safety or effectiveness of the device based on functional changes to the device’s operation (Note: modifications in the intended use of the device are covered in Deciding When to Submit a 510(k) for a Change to an Existing Device (http://www.fda.gov/downloads/MedicalDevices/DeviceRegulationandGuidance/GuidanceDocuments/ucm080243.pdf)). The impact of software changes on safety and effectiveness may not always be clear. This is often the case when making general code changes to software that are not necessarily intended to change function, but rather to perform what could be described as “code maintenance” or “infrastructure” modifications. These types of changes can, if not controlled properly, create unexpected changes to how the device functions. These types of changes, as well as others described in this section, should therefore involve a careful evaluation of their potential impact on device safety and effectiveness.
In addition to change management, these types of changes should also involve careful consideration of the overall architecture of the software. If the software architecture was developed in a planned, modular format, the likelihood of unintended impact to other areas of the code may be significantly reduced. On the other hand, if the software code was developed in a looser construct, without a clear architectural plan, the ability to clearly delineate between functional modules in the code may be reduced. The potential impact to device safety and effectiveness increases in code with looser construct, due to the inherent risk of unintended changes in code without clear boundaries in the functional modules.
The purpose of this section is to provide guidance regarding evaluation of certain types of software changes, such as “code maintenance” and “infrastructure” changes. Manufacturers are encouraged to discuss these “gray areas” with the relevant CDRH or CBER Office and Division if there are questions about whether to submit a new 510(k) for these or other types of software changes. In most cases, this will be the Division under which the device was originally cleared.
Common Software Change Types 🔗
The following list of common change types are intended to help manufacturers consider additional factors that may affect a decision to submit a new 510(k). Note that this list is not exhaustive. Any questions should be discussed with the respective CDRH Offices and/or CBER Offices and/or Divisions responsible for the device being modified.
Some of the common software change types include:
-
“Infrastructure” changes are modifications made to the software support system. Examples include but are not limited to: switching compilers, changing programming languages (C to C++, C++ to Java), or changing software drivers or libraries. The complexity of the change should be taken into consideration while determining whether the change requires submission of a new 510(k). For example, when changing programming languages, the similarity of the programming syntax between the two languages, as well as other factors (such as the coding paradigm associated with the old and new code), should be considered. A change from C to C++ may not entail significant code writing if the syntax is similar. On the other hand, moving from a functional or logical coding paradigm to an Object Oriented Programming paradigm, in conjunction with the change from C to C++, could involve significant software re-write, and submission of a new 510(k) is likely required.
Similar analysis generally applies to software driver changes, OS changes, etc. It should be noted that significant changes to verification and validation scripts might be a signal that significant infrastructure changes have taken place and should be examined. Updates to scripts alone do not indicate that submission of a new 510(k) is required; however, it is important to understand why the scripts are being updated.
- “Architecture” changes are modifications to the overall structure of the Examples include but are not limited to: porting to a new OS, software changes to support a new hardware platform, and new middleware. These changes may impact the overall performance of the device or extend the environment in which the device can operate. The extent of the changes and the impact that they have on the device should be considered in determining whether submission of a new 510(k) is required.
- “Core algorithm” changes are modifications made to an algorithm that directly impact or contribute to the device’s intended use. Examples include: alarm algorithms on a monitor, a motor control algorithm for an infusion pump, and a detection module and measurement engine algorithm for an IVD. Changes to the core algorithm that impact performance are addressed by the preceding section and flowchart. However, it is important to understand that a complete rewrite of the algorithm, even with the same performance claims and risk profile, may be significant enough to require submission of a new 510(k) because the rewrite may impact performance indirectly.
- “Clarification of Requirements – No Change to Functionality” are changes made to clarify software requirements after a product has received premarket clearance. This clarification may be revised phrasing of an existing requirement or creation of a new requirement altogether, without changing or adding functionality. Changes made to clarify the requirements as discussed here likely do not require submission of a new 510(k).
- “Cosmetic Changes – No Change to Functionality” are changes made to the appearance of the device that do not impact the clinical use of the device. For example, changing the company logo that is displayed on the background of every screen could involve modifying multiple software modules; while the number of modules impacted may be large, it is unlikely that the intended change could significantly impact the device’s safety and effectiveness or intended use, and submission of a new 510(k) is likely not required.
- “Reengineering” and “refactoring” are two common software maintenance techniques. “Reengineering” is defined as the examination and alteration of software to reconstitute it in a new form, and includes the subsequent implementation of the new form. It is often undertaken to replace aging legacy software. “Refactoring” is a disciplined technique for restructuring a software program’s internal structure without changing its clinical performance specification. Refactoring seeks to improve a program structure and its maintainability. In general, reengineering often results in broader and more complex changes, while refactoring is often narrower in scope and less complex. The complexity of the change and the impact on risk controls or performance should be considered to determine whether the change requires submission of a new 510(k). Changes that are minor modifications to enhance the maintainability of the device within its specification context are unlikely to require submission of a new 510(k). Changes involving significant software re-write likely require submission of a new 510(k) because of the impact on the performance and on the risk controls.
Appendix A. Software Modification Examples 🔗
The following are hypothetical examples of software changes with explanations as to why they likely would or would not require submission of a new 510(k). Note that these generalized examples do not necessarily account for every possible detail, risk, or consideration a manufacturer should evaluate, and should not be taken to mean that the changes described definitely do or do not require submission of a new 510(k). Real-world device modification decisions will depend on the particular details of the change and the specific device in question.
The examples below are only intended to illustrate the principles and recommendations discussed above with regard to a particular question. As such, the examples each contain only the response to the question that is being highlighted; this does not necessarily mean that an earlier question would not have appropriately led to a decision to submit a new 510(k).
1. Flowchart Question 1 Examples 🔗
1.1. Proactive software security patch 🔗
Description: A device manufacturer finds a security vulnerability as part of an ongoing security evaluation of their device. The manufacturer modifies the software solely to remove this vulnerability. The manufacturer’s analysis determined that the change does not have any other impact on the software or the device.
| # | Question | Yes/No | Rationale |
|---|---|---|---|
| 1 | Is the change made solely to strengthen cybersecurity and does not have any other impact on the software or device? | Yes | The change is made solely to address cybersecurity vulnerabilities or to strengthen cybersecurity. The manufacturer’s analysis determined that the change does not impact any other aspects of the software or device. |
Outcome: Document the change to file.
1.2. Adding encryption and additional access control for remote users 🔗
Description: A manufacturer makes a software modification to add encryption to the configuration file of the device, and add passcode requirements for remote users, in addition to the password needed to access the device. A timeout is also added for remote users. The manufacturer’s analysis determined that the change does not have any other impact on the software or the device.
| # | Question | Yes/No | Rationale |
|---|---|---|---|
| 1 | Is the change made solely to strengthen cybersecurity and does not have any other impact on the software or device? | Yes | The change is made to restrict user/customer access to appropriate levels and provide protection to the device configuration information, in order to strengthen the cybersecurity of the device. The manufacturer’s analysis determined that the change does not have any other impact on the software or the device. |
Outcome: Document the change to file.
2. Flowchart Question 2 Examples 🔗
2.1. Modify system to meet specification 🔗
Description: A manufacturer makes a software modification to prevent system software from truncating Specimen Identification (ID) barcode information. Without the change, the software system would truncate the Specimen ID from the point of an inserted invalid character. For instance, if the invalid character was “%” and the Specimen ID barcode was “12345%678,” the system software would read and assign a Specimen ID of “12345.” This defect could lead to mis-association of patient data. Incorrect software collation of patient information with patient results could lead to incorrect reports. The specification of the most recently cleared device indicated what constituted an invalid character and how invalid characters were to be handled. However, the software did not handle this one particular invalid character in line with the specification. A change is made to the software to prevent the truncation of Specimen ID barcode information where an invalid character has been inserted.
| # | Question | Yes/No | Rationale |
|---|---|---|---|
| 2 | Is the change made solely to return the system into specification of the most recently cleared device? | Yes | The software change disallowed use of the specific invalid character in Specimen IDs as defined in the instrument host interface specification. The original specification indicated how all illegal characters were to be handled. The existing device handled all but one as indicated in the specification. The change is made solely to ensure the software meets the original specification. |
Outcome: Document the change to file.
2.2. Correcting DICOM retrieve parameter error 🔗
Description: A PACS (Picture Archiving and Communication System) is able to automatically retrieve prior studies from a radiology information system to allow comparison with the current study. A software error resulted in a non DICOM-compliant (Digital Imaging and Communications in Medicine standard; http://dicom.nema.org/) sending of query parameters that prevented the automatic fetching of prior studies. A manual workaround existed, allowing the user to open these prior studies as needed. The manufacturer implements a software change to bring the product back to specification regarding DICOM conformance (send and retrieve).
| # | Question | Yes/No | Rationale |
|---|---|---|---|
| 2 | Is the change made solely to return the system into specification of the most recently cleared device? | Yes | The software change is implemented solely to return the system into specification of the most recently cleared device regarding DICOM conformance (send and retrieve) by automatically opening prior studies as expected in a routing reading workflow. |
Outcome: Document the change to file.
2.3. Error during maintenance procedure 🔗
Description: A manufacturer makes a software modification to fix an automated scheduled daily maintenance procedure. The defect concerned the cleaning solution bottle size parameter used in a maintenance procedure. The defect impacted the system’s ability to detect fluid on the bottle septum and caused intermittent fluid detection errors during the maintenance procedure. The user may need to repeat the procedure 2-3 times to complete the procedure without error. A software change is made to update the size parameter as was originally documented in the software specifications.
| # | Question | Yes/No | Rationale |
|---|---|---|---|
| 2 | Is the change made solely to return the system into specification of the most recently cleared device? | Yes | The change is to correct the software error to change the bottle size parameter back to the specified bottle size to bring system back into specification. |
Outcome: Document the change to file.
2.4. Data error 🔗
Description: An issue was observed in IVD analyzer software that collects reagent administrative records (e.g., material number, lot number, expiration date). The records are to be written by the software into a database table. After enough records are collected to fill the table, newly-collected records are then to be written in the first row of the table, overwriting previous records. Because of a software bug, the system mistakenly merges the new data with the existing data in the first row of the table. The cause of the anomaly was determined to be a coding error that did not affect any of the software requirements. A change was made to correct the software code in the control unit of the analyzer to ensure that data written to a row in the table is not merged with any existing data. The change to the software involved modification of a table within the analyzer software to add new columns to track the administrative data stored for reagents to prevent data from being merged.
| # | Question | Yes/No | Rationale |
|---|---|---|---|
| 2 | Is the change made solely to return the system into specification of the most recently cleared device? | Yes | The change was only to address a software anomaly and was not a change in specification or functionality of the most recently cleared device. |
Outcome: Document the change to file.
2.5. Database error 🔗
Description: An issue was observed for an IVD analyzer in the field. The IVD analyzer software collects reagent administrative records (e.g., material number, lot number, expiration date). The records are to be written by the software into a database table. After enough records are collected to fill the table, newly-collected records are then to be written in the first row of the table, overwriting previous records. Under certain conditions, the software system mistakenly merges the new data with the existing data in the first row of the table in the database, which may lead to an incorrect result. The cause of the bug was found to be an incorrectly worded software requirement that led to an error in the software code. The requirement was rewritten. An additional software change was made to correct the software code in the control unit of the analyzer. Code was modified to ensure that data written to a database is not merged with any existing data. The change to the software involved creating an entirely separate database within the instrument software, specifically for the administrative records stored for reagents to prevent records from being merged. This change required a specification change at the unit level to describe the new database.
| # | Question | Yes/No | Rationale |
|---|---|---|---|
| 2 | Is the change made solely to return the system into specification of the most recently cleared device? | No | A change was made to correct a coding error by adding a new database. This caused a change to the design specifications of the software. |
Outcome: Continue to question 3.
3. Flowchart Question 3a Examples 🔗
3.1. Adding a new diagnostic parameter 🔗
Description: An electroencephalogram (EEG) diagnostic monitor was cleared with spectral edge frequency (SEF) and peak power (PP) as quantitative parameters. The device’s intended use is to monitor brain electrical activity through electrodes placed on the surface of the head. A software modification is made to add Amplitude Integrated EEG (aEEG) as an additional quantitative parameter that was not included in the original premarket notification.
| # | Question | Yes/No | Rationale |
|---|---|---|---|
| 3a | Does the change introduce a new risk or modify an existing risk that could result in significant harm and that is not effectively mitigated in the most recently cleared device? | Yes | The hazardous situation most commonly associated with quantitative diagnostic parameters is the risk of incorrect or confusing information to the physician leading to a misdiagnosis, which could result in significant harm. While the causes of incorrect information for SEF and PP would be included in the original risk files, aEEG introduces a new cause related to an error in the aEEG calculation. Submission of a new 510(k) is required because the new cause is not effectively mitigated in the most recently cleared device and the hazardous situation, as discussed above, could result in significant harm. |
Outcome: Submit the change in a new 510(k).
3.2. Removing a diagnostic parameter 🔗
Description: An electroencephalogram (EEG) diagnostic monitor was cleared with Spectral Edge Frequency (SEF) and Peak Power (PP). SEF and PP are used by neurologists as quantitative parameters along with the raw EEG trace and other clinical metrics to arrive at a clinical decision. The device’s intended use is to monitor brain electrical activity through electrodes placed on the surface of the head. A modification is made to remove PP from the displayed quantitative parameters based on a marketing-conducted survey that indicated customers did not use PP in their clinical decisions.
| # | Question | Yes/No | Rationale |
|---|---|---|---|
| 3a | Does the change introduce a new risk or modify an existing risk that could result in significant harm and that is not effectively mitigated in the most recently cleared device? | No | Removal of PP does not introduce a new risk or modify any of the existing risks for the device. |
Outcome: Continue to question 3b.
3.3. Customer maintenance procedure 🔗
Description: The manufacturer makes a software modification to prevent a patient sample probe motor from overheating during a customer maintenance procedure. Power is applied to the sample probe motor to keep the sample probe assembly in a locked position during the user maintenance procedure. In the field, it was reported that applying power to the sample probe motor for more than 20 minutes causes the motor to overheat and creates a potential minor burn hazard (i.e., it becomes too hot to touch safely). The software change applies a timeout to power being applied to the sample probe motor during the maintenance procedure; after ten minutes, power to the sample probe motor is turned off. An additional software change adds a message window at the beginning of the procedure to alert the user that the procedure must be completed within a ten-minute window or the system will cut power to the motor. A limit of ten minutes was determined to keep the motor from overheating to the point of creating a potential minor burn hazard.
| # | Question | Yes/No | Rationale |
|---|---|---|---|
| 3a | Does the change introduce a new risk or modify an existing risk that could result in significant harm and that is not effectively mitigated in the most recently cleared device? | No | The change provides a mitigation to an existing hazardous situation that was not appropriately mitigated in the cleared device. However, the hazardous situation could not cause significant harm. |
Outcome: Continue to question 3b.
3.4. Adding new programming mode to a cardiac monitor 🔗
Description: The device is an implantable, automatically activated monitoring system that records subcutaneous electrocardiograms designed to record the arrhythmias in a patient. The manufacturer has made a software modification to add an alternative programming mode to change the way the device interacts with the programmer. This new programming mode provided different capabilities for data programming, interrogating, and managing the device data and function. The mode introduces new technology that impacts the safety profile of the device as a result of the energy transfer that occurs during programming.
Outcome: Submit the change in a new 510(k).
| # | Question | Yes/No | Rationale |
|---|---|---|---|
| 3a | Does the change introduce a new risk or modify an existing risk that could result in significant harm and that is not effectively mitigated in the most recently cleared device? | Yes | This feature introduces new risks based on the new programming mode that could cause significant harm as a result of energy transfer to the patient. |
3.5. Imaging catheters – new optical module and new laser 🔗
Description: The device is an imaging catheter for coronary arteries that includes lasers and optical components. The manufacturer modifies the device software to integrate new optical modules and a new advanced laser method. The integration of the new components and function pose new risks to patients as a result of the new control parameters for the laser which could result in patient injury if not integrated appropriately.
| # | Question | Yes/No | Rationale |
|---|---|---|---|
| 3a | Does the change introduce a new risk or modify an existing risk that could result in significant harm and that is not effectively mitigated in the most recently cleared device? | Yes | The change introduces new hazardous situations associated with interoperability. This change introduces a new hazardous situation as a result of the optical module not recognizing the new catheter and therefore not providing the correct laser settings, which could result in significant harm. |
Outcome: Submit the change in a new 510(k).
4. Flowchart Question 3b Examples 🔗
4.1. Modification of a risk control 🔗
Description: The device is a robotically assisted surgical system that utilizes position sensors. The system incorporates primary and secondary sensors to monitor the movement of actuators to prevent uncontrolled motion of the instrument in the event of a component failure. The system goes into a fault state and halts motion if the position information between the sensors does not match within a certain threshold. The threshold for each actuator is programmed in the software and there is a specification for how much overall movement is acceptable at the tip of the instrument before movement stops. The manufacturer makes a software change to the threshold settings for the position sensors; specifically, the software specification that defines the tip movement was widened and the software was changed to allow the wider tolerance. The change was made to minimize false assertion of the safety system, and the change in the specification for movement at the tip of the instrument was still within an appropriate safety tolerance for the device, as determined by analysis done by the manufacturer. However, the change modified an existing risk control (distance that can be traveled under fault conditions) that could significantly affect safety or effectiveness.
| # | Question | Yes/No | Rationale |
|---|---|---|---|
| 3a | Does the change create or necessitate a new risk control measure or a modification of an existing risk control measure for a hazardous situation that could result in significant harm? | Yes | The modified threshold values do not meet the specification for overall tip movement, which was required in the most recently cleared device to effectively mitigate the hazardous situation that could result in significant harm. Thus, the change necessitated modification of an existing risk control in the most recently cleared device and submission of a new 510(k) is required. |
Outcome: Submit the change in a new 510(k).
4.2. Modification of threshold settings 🔗
Description: The device is a robotically assisted surgical system that utilizes position sensors. The system incorporates primary and secondary sensors to monitor the movement of actuators to prevent uncontrolled motion of the instrument in the event of a component failure. The system goes into a fault state and halts motion if the position information between the sensors does not match within a certain threshold. The threshold for each actuator is programmed in the software and there is a specification for how much overall movement is acceptable at the tip of the instrument before movement stops. The manufacturer makes a software change to the threshold settings for the position sensors; specifically, the software was modified to better calculate overall movement. The change was made to minimize false assertion of the safety system, which required the surgeon to hit an override button to continue. This requirement can be a nuisance and distract from surgery. The modified software continued to meet the specification for movement at the tip of the instrument after a component failure.
| # | Question | Yes/No | Rationale |
|---|---|---|---|
| 3b | Does the change create or necessitate a new risk control measure or a modification of an existing risk control measure for a hazardous situation that could result in significant harm? | No | This change modifies sensor threshold parameters so that transient conditions that can be present during normal operation do not cause unnecessary activation of the risk control measure. The change makes the system more noise-tolerant without impacting true positive detection for the risk control measure. The overall movement criteria are met under all fault conditions. |
Outcome: Continue to Question 4.
4.3. Adding user interface alerts and controls 🔗
Description: An IVD analyzer manufacturer makes software modifications to replace existing modes of controls for handling samples having invalid characters in specimen IDs (specimen identification mis-association) received from Laboratory Information System or middleware vendors. Existing manual modes of control were adequate, but required operator interaction to evaluate whether a result record for a sample had an invalid specimen ID. The new modes of control include additional automation through a design improvement that will not generate results for a sample having an invalid specimen ID. Instead, the system software will: (1) generate a warning message to the operator that an invalid specimen ID was detected; (2) not generate or report results for a sample having an invalid specimen ID; and (3) create a system log entry.
| # | Question | Yes/No | Rationale |
|---|---|---|---|
| 3b | Does the change create or necessitate a new risk control measure or a modification of an existing risk control measure for a hazardous situation that could result in significant harm? | Yes | This software change modifies the risk control that identifies invalid characters by automating a previously manual process. If the invalid characters are not identified appropriately, then patient laboratory test results could be lost or replaced by incorrect results. Loss or replacement of results could influence treatment decisions, which could cause significant harm. |
Outcome: Submit the change in a new 510(k).
4.4. Print patient information on PACS report 🔗
Description: A PACS provides the option to print images along with a copy of the diagnostic findings from the radiologist. There is data on each page allowing the user to match each page to the corresponding information (e.g., patient ID, Study Identifier). This data helps to address the known risk of pages being mixed-up after printout. Based on customer preference, the manufacturer decided to enhance this existing risk control and have actual patient information and demographics printed on each page. This is intended to be easier for the user to identify which pages belong together and, as a result, further decrease the risk of mixing up printed pages.
| # | Question | Yes/No | Rationale |
|---|---|---|---|
| 3b | Does the change create or necessitate a new risk control measure or a modification of an existing risk control measure for a hazardous situation that could result in significant harm? | Yes | The risk is already sufficiently mitigated with the original risk controls (that is, to have patient identification related information on each printed page). This software modification is a redundant risk control that was not made in response to a new, modified, or previously unknown hazardous situation or cause thereof. |
Outcome: Continue to Question 4.
4.5. Infusion pump alarm 🔗
Description: A general purpose infusion pump has one alarm to alert the user when an occlusion has been detected. The software change modifies the existing alarm to provide two alarms related to occlusion: occlusion downstream and occlusion upstream. These alarms provide specific information to help resolve the occlusion.
| # | Question | Yes/No | Rationale |
|---|---|---|---|
| 3b | Does the change create or necessitate a new risk control measure or a modification of an existing risk control measure for a hazardous situation that could result in significant harm? | Yes | The change modifies the risk control, i.e., the alarm, which is already present for occlusion. This risk control is necessary to improve safety by effectively mitigating specific occlusion events that could result in significant harm if not resolved correctly. |
Outcome: Submit the change in a new 510(k).
5. Flowchart Question 4 Examples 🔗
5.1. Improve sample throughput 1 🔗
Description: A manufacturer makes a software performance enhancement to improve sample throughput time by 20%. Software modifications include changes to decrease assay cycle times by allowing for shorter sample reaction incubation times. Decreasing sample assay times could have an impact on run performance and/or assay performance in a manner that could have a negative impact on diagnosis or therapy delivered to patients.
| # | Question | Yes/No | Rationale |
|---|---|---|---|
| 4 | Could the change significantly affect clinical functionality or performance specifications that are directly associated with the intended use of the device? | Yes | The change is to increase the throughput performance specification, but has a significant impact on the performance of the device. There is a shorter reaction incubation time and therefore a potential significant impact on diagnostic utility and effectiveness. |
Outcome: Submit the change in a new 510(k).
5.2. Improve sample throughput 2 🔗
Description: A manufacturer makes a software modification to improve sample throughput by 5% by decreasing pre-analytic processing time. Software modifications include a change to decrease sample delivery time from the sample load area to the sample aspiration area. As described here, decreasing sample delivery times do not have an impact on assay performance.
| # | Question | Yes/No | Rationale |
|---|---|---|---|
| 4 | Could the change significantly affect clinical functionality or performance specifications that are directly associated with the intended use of the device? | No | The modifications do not impact assay performance as it relates to intended use. Improvement resulted from technical analysis of the sample delivery algorithm to optimize timing and remove unnecessary timing delays. |
Outcome: If the factors identified in Section VI are not relevant for this change, document the change to file.
5.3. Software change to modify summary window 🔗
Description: A manufacturer makes a software modification to increase the number of images that can be viewed in a summary view for an ingestible telemetric gastrointestinal capsule imaging system. The new software allows for four images to be viewed simultaneously instead of two while a user reviews the images. The specifications for the image quality are not impacted by this change.
| # | Question | Yes/No | Rationale |
|---|---|---|---|
| 4 | Could the change significantly affect clinical functionality or performance specifications that are directly associated with the intended use of the device? | No | The change does not significantly impact functionality or performance specifications that are directly associated with the intended use of the device. Having more images in the window allows for the physician to review more images without increasing software loading time. |
Outcome: If the factors identified in Section VI are not relevant for this change, document the change to file.
5.4. OEM module 🔗
Description: A multi-parameter monitor device was originally cleared with version A of an original equipment manufacturer (OEM) module for blood oxygen saturation (SpO2). The OEM makes a change to version A of the SpO2 sensor. The change to the SpO2 sensor did not require submission of a new 510(k) and the change did not impact the specifications for the SpO2 in terms of data acquisition or processing. However, the SpO2 does identify itself to the multi-parameter monitor using a new version number. This requires a software change on the multi-parameter monitor to allow for successful interoperability with the new version of the sensor.
| # | Question | Yes/No | Rationale |
|---|---|---|---|
| 4 | Could the change significantly affect clinical functionality or performance specifications that are directly associated with the intended use of the device? | No | The clinical functionality is not affected. The software modification allows for successful integration of this version of the sensor. |
Outcome: If the factors identified in Section VI are not relevant for this change, document the change to file.
5.5. Sterilizer user interface change 🔗
Description: A sterilizer display provides vital information on the temperature, the pressure, and the remaining cycle time. Software changes are made to increase the font size of these parameters on the display due to customer feedback (not related to any adverse events). The items are all in the same location and the appearance is unchanged aside from the larger font size.
| # | Question | Yes/No | Rationale |
|---|---|---|---|
| 4 | Could the change significantly affect clinical functionality or performance specifications that are directly associated with the intended use of the device? | No | Since the information was previously displayed, the change has no significant effect on the functionality or the performance of the device. |
Outcome: If the factors identified in Section VI are not relevant for this change, document the change to file.
5.6. Modify device algorithms 🔗
Description: A manufacturer makes a software modification to enhance an arrhythmia detection algorithm. The device is intended to provide detection alarms for life- threatening arrhythmias in an intensive care unit (ICU) environment. The change impacts sensitivity and specificity and therefore the detection of arrhythmias, which are critical to the clinical performance of the device.
| # | Question | Yes/No | Rationale |
|---|---|---|---|
| 4 | Could the change significantly affect clinical functionality or performance specifications that are directly associated with the intended use of the device? | Yes | The modification has direct impact on diagnostic performance of the device in that the performance of the arrhythmia detection was changed. |
Outcome: Submit the change in a new 510(k).
5.7. Modification to alarm duration 🔗
Description: A manufacturer makes a software modification to allow users to silence a low-risk alarm on a dialysis system. The change consists of a “snooze” button that silences the alarm for a set amount of time before resounding.
| # | Question | Yes/No | Rationale |
|---|---|---|---|
| 4 | Could the change significantly affect clinical functionality or performance specifications that are directly associated with the intended use of the device? | No | The silencing of a non-critical alarm does not impact the clinical functionality. The criteria for the alarm are unchanged from the most recently cleared device. |
Outcome: If the factors identified in section VI are not relevant for this change, document the change to file.
Footnotes 🔗
-
IMDRF/SaMD WG/N10: Software as a Medical Device (SaMD): Key Definitions (http://www.imdrf.org/docs/imdrf/final/technical/imdrf-tech-131209-samd-key-definitions-140901.pdf). ↩
-
This guidance applies to devices granted marketing authorization via the De Novo classification process and that are not exempt from premarket notification requirements. ↩
