
Innolitics introduction 🔗
In November of 2021, the FDA released a draft guidance document titled Content of Premarket Submissions for Device Software Functions. This draft guidance will replace the 2005 guidance document titled Guidance for the Content of Premarket Submissions for Software Contained in Medical Devices. We've transcribed the PDF draft guidance into HTML here so that we can send our clients links to particular sections of the document.
If you're overwhelmed by cybersecurity requirements for your SaMD, we can help. We provide turn-key engineering solutions and regulatory consulting services. Contact us if you'd like a team that help bring your device to the market as quickly as possible. If you have an engineering team, we can also integrate cybersecurity into your quality system.
Thank you to everyone at the FDA for their efforts to keep patients safe and make device manufacturer's jobs as easy as possible through these documents.
About this Transcript 🔗
This document is a transcript of an official FDA (or IMDRF) guidance document. We transcribe the official PDFs into HTML so that we can share links to particular sections of the guidance when communicating internally and with our clients. We do our best to be accurate and have a thorough review process, but occasionally mistakes slip through. If you notice a typo, please email a screenshot of it to Mihajlo at mgrcic@innolitics.com so we can fix it.
Preamble 🔗
This draft guidance was issued on November 4, 2021.
For questions about this document regarding CDRH-regulated devices, contact the Digital Health Center of Excellence at digitalhealth@fda.hhs.gov. For questions about this document regarding CBER regulated devices, contact the Office of Communication, Outreach, and Development (OCOD) at 1-800-835-4709 or 240-402-8010, or by email at ocod@fda.hhs.gov.
Contains Nonbinding Recommendations
I. Introduction 🔗
This guidance document is intended to provide information regarding the recommended documentation sponsors should include in premarket submissions for FDA’s evaluation of the safety and effectiveness of device software functions, which are functions that meet the definition of a device under section 201(h) of the Federal Food, Drug, and Cosmetic Act (FD&C Act).1 The recommendations in this guidance document pertain to device software functions, including software in a medical device (SiMD) and software as a medical device (SaMD).2 When final, this document will replace FDA’s Guidance for the Content of Premarket Submissions for Software Contained in Medical Devices3 issued on May 11, 2005, and it will update FDA’s thinking related to the documentation FDA recommends sponsors include for the review of device software functions in premarket submissions. This guidance identifies the software information generally necessary for evaluating the safety and effectiveness of a device in a premarket submission. The recommendations in this guidance also may help facilitate FDA’s premarket review. This guidance describes information that would be typically generated and documented4 during software development, verification, and design validation. The least burdensome approach was applied to identify the minimum amount of information that, based on our experience, would generally be needed to support a premarket submission for a device that uses software. During premarket review, FDA may request additional information that is needed to evaluate the submission. For example, in order to demonstrate a reasonable assurance of safety and effectiveness for devices that use software, documentation related to the requirements of the Quality System Regulation (QSR) (21 CFR Part 820) is often a necessary part of the premarket submission. As part of QSR design controls, a manufacturer must “establish and maintain procedures for validating the devices design,” which “shall include software validation and risk analysis, where appropriate.” (21 CFR 820.30(g)). The documentation recommended in this guidance is based on FDA’s experience evaluating the safety and effectiveness of device software. However, sponsors may use alternative approaches and provide different documentation so long as their approach and documentation satisfies premarket submission requirements in applicable statutory provisions and regulations. For the current edition of the FDA-recognized consensus standard(s) referenced in this document, see the FDA Recognized Consensus Standards Database.5 For more information regarding use of consensus standards in regulatory submissions, please refer to the FDA guidance titled Appropriate Use of Voluntary Consensus Standards in Premarket Submissions for Medical Devices6 and Standards Development and the Use of Standards in Regulatory Submissions Reviewed in the Center for Biologics Evaluation and Research7 The contents of this document do not have the force and effect of law and are not meant to bind the public in any way, unless specifically incorporated into a contract. This document is intended only to provide clarity to the public regarding existing requirements under the law. FDA guidance documents, including this guidance, should be viewed only as recommendations, unless specific regulatory or statutory requirements are cited. The use of the word should in Agency guidance means that something is suggested or recommended, but not required.
II. Background 🔗
The purpose of this guidance is to describe FDA’s thinking on the recommended documentation sponsors should include in premarket submissions for FDA’s evaluation of the safety and effectiveness of device software functions. This thinking recognizes recent changes to the FD&C Act made by the 21st Century Cures Act (Cures Act), which amended section 520 of the FD&C Act and excludes certain software functions from the device definition. It also considers the rapidly evolving nature of digital health and recent FDA recognized consensus standards related to software. This guidance, as described in Section III (Scope), is intended to complement other existing guidance documents that provide recommendations related to software, including the guidance documents listed below. The following guidance documents represent a subset of FDA guidances with digital health content8 relevant to premarket software documentation activities. Please note the list is not exhaustive and is subject to change:
- Multiple Function Device Products: Policy and Considerations9
- Off-The-Shelf Software Use in Medical Devices10,11
- Design Considerations and Premarket Submission Recommendations for Interoperable Medical Devices12
- General Principles of Software Validation13
- Content of Premarket Submissions for Management of Cybersecurity in Medical Devices14
- Cybersecurity for Networked Medical Devices Containing Off-The-Shelf (OTS) Software15
FDA encourages the consideration of these guidances when developing device software functions and preparing premarket software documentation.
The emergence of consensus standards related to software has helped to improve the consistency and quality of software development and documentation, particularly with respect to activities such as risk assessment and management. When possible, FDA harmonized the terminology and recommendations in this guidance with software-related consensus standards, such as the following examples. The following standards are not intended to represent an exhaustive list and are subject to change:
- ANSI/AAMI/ISO 14971: Medical devices - Applications of risk management to medical devices
- ANSI/AAMI/IEC 62304: Medical Device Software - Software Life Cycle Processes (also referred to as “IEC 62304” in this guidance)
- ANSI/AAMI SW91: Classification of defects in health software
The Agency encourages the consideration of these FDA recognized consensus standards when developing device software functions and preparing premarket software documentation.
III. Scope 🔗
For the purposes of this document, FDA refers to a software function that meets the definition of a device as a “device software function.” For example, a device software function may control a hardware device or be part of a hardware device16 (i.e., Software in a Medical Device, or SiMD) or be a device without being part of a hardware device17 (i.e., Software as a Medical Device, or SaMD).18 For any given product, the term “function” is a distinct purpose of the product, which could be the intended use or a subset of the intended use of the product. For example, a product with an intended use to analyze data has one function: analysis. A product with an intended use to store, transfer, and analyze data has three functions: (1) storage, (2) transfer, and (3) analysis. As this example illustrates, a product may contain multiple functions.
This guidance is intended to cover:
- firmware and other means for software-based control of medical devices;
- stand-alone software applications;
- software intended to be operated on general-purpose computing platforms;
- dedicated hardware/software medical devices; and
- accessories to medical devices when those accessories contain or are composed of software.
This guidance applies to all types of premarket submissions that include one or more device software function(s). Premarket submissions include:
- Premarket Notification (510(k));
- De Novo Classification Request;
- Premarket Approval Application (PMA);
- Investigational Device Exemption (IDE);
- Humanitarian Device Exemption (HDE); and
- Biologics License Application (BLA).
This guidance does not apply to automated manufacturing and Quality System software19 or software that is not a device. For further information or to clarify the documentation expectations, please contact the responsible FDA review division.
Generally, the recommendations in this guidance apply to the device constituent part of a combination product20 (such as drug-device and biologic-device combination products) when the
device21 constituent part includes a device software function. For more information, contact the FDA review division that will have the lead review for the combination product.
Other FDA guidance documents may recommend additional software-related documentation for inclusion in a premarket submission. For example, recommendations regarding cybersecurity information to include in a device premarket submission can be found in the guidances Content of Premarket Submissions for Management of Cybersecurity in Medical Devices and Cybersecurity for Networked Medical Devices Containing Off-The-Shelf (OTS) Software.22 Section II (Background) references other relevant guidance documents that supplement the
recommendations contained in this guidance.
Device software functions subject to specific special controls may require additional software related documentation in a premarket submission. As applicable, please refer to the relevant special controls for the device.
This guidance does not apply to the software-related documentation that may be needed to evaluate postmarket software device issues, including corrections and removals.
While this guidance identifies the documentation sponsors should include in premarket submissions, this guidance is not meant to provide recommendations regarding how device software should be developed, verified and validated. For recommendations on device software development, verification and validation, please consult software-related FDA recognized
consensus standards and other software-related FDA guidance documents mentioned in this guidance (e.g., General Principles of Software Validation23).
IV. Definitions 🔗
The following terms are used for the purposes of this guidance:
A. Device Software Function
Software function that meets the device definition in section 201(h) of the FD&C Act. “Software as a Medical Device (SaMD)” and “Software in a Medical Device (SiMD)”24 are device software functions. As discussed above, the term “function” is a distinct purpose of the product, which could be the intended use or a subset of the intended use of the product.
B. Off-the-Shelf Software25,26
A generally available software component used by a device manufacturer for which the manufacturer cannot claim complete software life cycle control (e.g., operating system, printer/display libraries).
C. Serious Injury27
An injury or illness that:
- Is life threatening,
- Results in permanent impairment of a body function or permanent damage to a body structure, or
- Necessitates medical or surgical intervention to preclude permanent impairment of a body function or permanent damage to a body structure. Permanent is defined as irreversible impairment or damage to a body structure or function, excluding trivial impairment or damage.
D. Software as a Medical Device (SaMD)28,29
Software that meets the definition of a device in section 201(h) of the FD&C Act and is intended to be used for one or more medical purposes without being part of a hardware device.30
E. Software in a Medical Device (SiMD)31
Software that meets the definition of a device in section 201(h) of the FD&C Act, and is used to control a hardware device or is necessary for a hardware device to achieve its intended use. Typically, SiMD is embedded within or is part of a
hardware device.32
F. Software Verification and Software Validation
This guidance uses the terms “software verification” and “software validation,” which are described in further detail below.
- For the purposes of this guidance, software verification is confirmation by objective evidence that the output of a particular phase of development meets all the input requirements for that phase. Software verification involves evaluating the consistency, completeness, and correctness of the software and its supporting documentation, as it is being developed, and provides support for a subsequent conclusion that software is validated. Software testing is one of several verification activities intended to confirm that the software development output meets its input requirements. Other verification activities include walk-throughs, various static and dynamic analyses, code and document inspections, module level testing and integration testing. For example, the input and output of the design phase are known as Software Requirements Specification (SRS) and Software Design Specification (SDS), respectively. In this case, software verification would involve confirming by objective evidence (e.g., reviews, traceability analysis) that the software design as documented in the SDS (i.e., output) correctly and completely implements all the requirements of the SRS (i.e., input).
- For the purposes of this guidance, software validation refers to establishing, by objective evidence, that the software specifications conform to user needs and intended uses, and that the particular requirements implemented through software can be consistently fulfilled. Software validation is a part of design validation of the finished device. It involves checking for proper operation of the software in its actual or simulated use environment, including integration into the final device where appropriate. Software validation is highly dependent upon comprehensive software testing and other verification tasks previously completed at each stage of the software development life cycle. Planning, requirements, traceability, testing, risk assessment, design reviews, change management, and many other aspects of good software engineering are important activities that together help to support a conclusion that software is validated.
The above descriptions of software verification and software validation are consistent with FDA’s thinking as described in the guidance General Principles of Software Validation.33
V. Documentation Level 🔗
The recommended documentation for a premarket submission depends on the device’s risk to a patient, a user of a device, or others in the environment of use. FDA intends to consider four risk-based factors to help determine the device’s Documentation Level, which is either Basic or Enhanced. The purpose of the Documentation Level is to help identify the minimum amount of information that would support a premarket submission that includes device software functions.
The Documentation Level is based on the device’s intended use.34 Evidence relevant to intended use may include the design of the device. Notably, the Documentation Level is determined by the intended use of the device as a whole—not the individual device function(s).
Basic Documentation should be provided for any premarket submission that includes device software functions where Enhanced Documentation does not apply.
Enhanced Documentation should be provided for any premarket submission that includes device software functions, where any of the following factors apply:
- The device is a constituent part of a combination product.35
- The device (a) is intended to test blood donations for transfusion-transmitted infections; or (b) is used to determine donor and recipient compatibility; or (c) is a Blood Establishment Computer Software.36
- The device is classified as class III.37
- A failure or latent flaw of the device software function(s) could present a probable risk of death or serious injury,38 either to a patient, user of the device, or others in the environment of use. These risk(s) should be assessed prior to implementation of risk control measures. You should consider the risk(s) in the context of the device’s intended use; the direct and indirect impacts to safety, treatment, and/or diagnosis; and other relevant considerations.
The term "probable" in factor 4 above is intended to capture reasonably foreseeable software and hardware risks associated with the device, including those risks resulting from intentional or reasonably foreseeable misuse of the device, prior to the implementation of risk control measures. “Probable” risks also include the likelihood that device functionality is intentionally or unintentionally compromised by inadequate device cybersecurity. The term “probable” is intended to exclude the consideration of purely hypothetical risks, consistent with the use of “probable” in other FDA guidances.39 It is the sponsor’s responsibility to proactively and comprehensively consider risks as part of the device’s risk assessment.
For additional information and examples on assessing the Documentation Level for a device, please refer to Appendix A.
VI. Recommended Documentation 🔗
This section reflects FDA’s recommendations for information to be included in premarket submissions for Basic and Enhanced Documentation Levels. This recommended information should demonstrate that practices were employed resulting in traceability for device software functions and demonstrate planning, requirements, risk assessment, design reviews, change management, testing plans and results, and other aspects of good software engineering for device software functions, to help inform a regulatory decision on whether the software is appropriately designed, verified, and validated.
Table 1 below provides an outline of the recommended documentation for each software documentation element and corresponding Documentation Level. Please refer to subsections A-J in this section of the guidance (IV) for more detail.
Table 1. Outline of Recommended Documentation
| Software Documentation Elements | Basic Documentation Level | Enhanced Documentation Level |
|---|---|---|
| Documentation Level Evaluation (Section IV.A) | A statement indicating the appropriate Documentation Level and a description of the rationale for that level. | A statement indicating the appropriate Documentation Level and a description of the rationale for that level. |
| Software Description (Section IV.B) | Software description, including overview of operationally significant software features, analyses, inputs, and outputs. | Software description, including overview of operationally significant software features, analyses, inputs, and outputs. |
| System and Software Architecture Design Chart (Section IV.C) | Detailed diagrams of the modules, layers, and interfaces that comprise the device, their relationships, the data inputs/outputs and flow of data, and how users or external products (including IT infrastructure and peripherals) interact with the system and software. | Detailed diagrams of the modules, layers, and interfaces that comprise the device, their relationships, the data inputs/outputs and flow of data, and how users or external products (including IT infrastructure and peripherals) interact with the system and software. |
| Risk Management File (Section IV.D) | Risk management plan, risk assessment demonstrating that risks have been appropriately mitigated, and risk management report. | Risk management plan, risk assessment demonstrating that risks have been appropriately mitigated, and risk management report. |
| Software Requirements Specification (SRS) (Section IV.E) | The complete documentation, describing the needs or expectations for a system or software, presented in an organized format and with sufficient information to understand the traceability of the information with respect to the other software documentation elements (e.g., risk management file, software design specification, system and software architecture design chart, software testing as part of verification and validation). | The complete documentation, describing the needs or expectations for a system or software, presented in an organized format and with sufficient information to understand the traceability of the information with respect to the other software documentation elements (e.g., risk management file, software design specification, system and software architecture design chart, software testing as part of verification and validation). |
| Software Design Specification (SDS) (Section IV.F) | None. | The complete documentation, including sufficient information that would allow FDA to understand the technical design details of how the software functions, how the software design completely and correctly implements all the requirements of the SRS and how the software design traces to the SRS in terms of intended use, functionality, safety, and effectiveness. |
| Software Development and Maintenance Practices (Section IV.G) | A Declaration of Conformity to IEC 62304 OR a summary of the life cycle development plan and a summary of configuration management and maintenance activities. |
A Declaration of Conformity to IEC 62304 OR Basic Documentation Level PLUS complete configuration management and maintenance plan document(s) |
| Software Testing as Part of Verification and Validation (Section IV.H) | A summary description of the testing activities at the unit, integration and system levels. System level test protocol including expected results, observed results, pass/fail determination, and system level test report. | Basic Documentation Level PLUS unit and integration level test protocols including expected results, observed results, pass/fail determination, and unit and integration level test reports. |
| Revision Level History (Section IV.I) | Revision history tabulating the major changes to the software during the development cycle, including date, version number, a brief description of the changes relative to the previous version, and indication of the version on which testing was performed. | Revision history tabulating the major changes to the software during the development cycle, including date, version number, a brief description of the changes relative to the previous version, and indication of the version on which testing was performed. |
| Unresolved Anomalies (e.g., Bugs, Defects, or Errors) (Section IV.J) | List of remaining software anomalies (e.g., bugs, defects) annotated with an explanation of the impact on safety or effectiveness, including operator usage and human factors, work-arounds, and timeframe for correction. | List of remaining software anomalies (e.g., bugs, defects) annotated with an explanation of the impact on safety or effectiveness, including operator usage and human factors, work-arounds, and timeframe for correction. |
<table class='table'>
<tr>
<th>Software Documentation Elements</th>
<th>Basic Documentation Level</th>
<th>Enhanced Documentation Level</th>
</tr>
<tr>
<th>Documentation Level Evaluation (Section IV.A)</th>
<td colspan=2>A statement indicating the appropriate Documentation Level and a description of the rationale for that level.</td>
</tr>
<tr>
<th>Software Description (Section IV.B)</th>
<td colspan=2>Software description, including overview of operationally significant software features, analyses, inputs, and outputs.</td>
</tr>
<tr>
<th>System and Software Architecture Design Chart (Section IV.C)</th>
<td colspan=2>Detailed diagrams of the modules, layers, and interfaces that comprise the device, their relationships, the data inputs/outputs and flow of data, and how users or external products (including IT infrastructure and peripherals) interact with the system and software.</td>
</tr>
<tr>
<th>Risk Management File (Section IV.D)</th>
<td colspan=2>Risk management plan, risk assessment demonstrating that risks have been appropriately mitigated, and risk management report.</td>
</tr>
<tr>
<th>Software Requirements Specification (SRS) (Section IV.E)</th>
<td colspan=2>The complete documentation, describing the needs or expectations for a system or software, presented in an organized format and with sufficient information to understand the traceability of the information with respect to the other software documentation elements (e.g., risk management file, software design specification, system and software architecture design chart, software testing as part of verification and validation).</td>
</tr>
<tr>
<th>Software Design Specification (SDS) (Section IV.F)</th>
<td>None</td>
<td>The complete documentation, including sufficient information that would allow FDA to understand the technical design details of how the software functions, how the software design completely and correctly implements all the requirements of the SRS and how the software design traces to the SRS in terms of intended use, functionality, safety, and effectiveness.</td>
</tr>
<tr>
<th>Software Development and Maintenance Practices (Section IV.G)</th>
<td>A Declaration of Conformity to IEC 62304<br> <b>OR</b><br> a summary of the life cycle development plan and a summary of configuration management and maintenance activities.</td>
<td>A Declaration of Conformity to IEC 62304<br> <b>OR</b><br> <b>Basic Documentation Level PLUS</b> complete configuration management and maintenance plan document(s)</td>
</tr>
<tr>
<th>Software Testing as Part of Verification and Validation (Section IV.H)</th>
<td>A summary description of the testing activities at the unit, integration and system levels. System level test protocol including expected results, observed results, pass/fail determination, and system level test report.</td>
<td><b>Basic Documentation Level PLUS</b> unit and integration level test protocols including expected results, observed results, pass/fail determination, and unit and integration level test reports.</td>
</tr>
<tr>
<th>Revision Level History (Section IV.I)</th>
<td colspan=2>Revision history tabulating the major changes to the software during the development cycle, including date, version number, a brief description of the changes relative to the previous version, and indication of the version on which testing was performed.</td>
</tr>
<tr>
<th>Unresolved Anomalies (e.g., Bugs, Defects, or Errors) (Section IV.J)</th>
<td colspan=2>List of remaining software anomalies (e.g., bugs, defects) annotated with an explanation of the impact on safety or effectiveness, including operator usage and human factors, work-arounds, and timeframe for correction.</td>
</tr>
</table>
A. Documentation Level Evaluation 🔗
A statement indicating the Documentation Level for the device and a description of the rationale for such Documentation Level, as appropriate.
B. Software Description 🔗
An overview of operationally significant software features, including images, flow charts, and state diagrams as needed to adequately explain the software functionality40 should be provided. If the premarket submission is for a modified device, provide the document number of the previous submission and highlight pertinent software changes since the last FDA approval or clearance.
Please consider and provide information to address the questions below when preparing the software description. However, FDA recognizes that these questions and examples may not capture all the unique aspects of a device software function and encourages the inclusion of additional information that will streamline or further FDA’s understanding of the device’s functionality to facilitate the review of a submission.
If the device is a multiple function device product and includes software function(s) that are considered “other functions,” as that term is used in the guidance Multiple Function Device Product: Policy and Considerations, the recommendations described in the aforementioned guidance should be considered when preparing the software description information.
Software Specifics
- What programming languages and compiler versions are used?
- What hardware platforms are used?
- What operating systems are used (if applicable)?
- Does the device use Off-The-Shelf (OTS) software(s)?41
- What is the intended release version? If the intended release version is different from the documentation’s version, explain the differences.
Software Operation
- Who operates the software (user)? The patient, a caregiver, a healthcare professional, or a combination thereof?
- What is the intended patient population?
- Does the software function focus on a specific disease, condition, patient characteristic or demographic?
- Does the software provide information that is directly applicable to a specific disease or condition?
- If the software performs an analysis of data, what is the analysis methodology? What is the evidence base used for this methodology? Examples: rule-based calculations, online test administration, artificial intelligence (AI)/machine learning (ML), neural networks, fixed or adaptive algorithms.
- Does the software impact or replace any otherwise manual or clinician performed actions? What are the workflow steps and assumptions (from beginning to end state)? Examples: automates steps, triages patients, provides a definite diagnosis or suggests likely diagnosis for further confirmation by physician, performs or recommends treatment, identifies a region of interest for further review.
- If the device is AI/ML-enabled, what populations or samples have informed the model(s)? What steps were taken to identify and address potential biases and limitations of the model(s)?
Software Inputs and Outputs
- What are the inputs and their format? Examples: data, images (specify modality), measurements (specify units), sensor/attachments, report, questionnaire.
- Who or what provides the inputs? Examples: user, other medical devices, other non medical devices or software.
- Is the device designed to be interoperable?42 In other words, does the device transmit, exchange, and/or use information through an electronic interface with another medical/nonmedical product, system, or device? If yes, what other products does the device interface with, and what methods, standards, and specifications are used to interact and/or communicate with other medical/nonmedical product, system, or device?
- What are the outputs and their format?
- Note: The performance testing of the outputs, including test setup, acceptance criteria, and results should be included in the submission. Please clearly indicate where in the submission this information is located. Examples: testing for accuracy and repeatability of output measurements, parametric analyses, model outputs, device generated segmentation contours, medical image enhancements.
- To whom are the outputs provided? Examples: patients, caregivers, healthcare professionals, technicians, researchers, health records, interoperable systems.
- What is the data or information flow of the software? Consider identifying the response to this question in the system and software architecture diagram. Examples: inputs or outputs transmitted locally, via cloud storage, by disk or drive, or wirelessly.
- Does the software interact with any networked devices? Does the software use cloud or network storage?
If any of the information requested above is included in another document, such as the SRS, an annotation and a reference to the document in the submission where this information is located should be provided.
C. System and Software Architecture Diagram 🔗
The purpose of the system and software architecture diagram is to present a roadmap of the device design to facilitate a clear understanding of 1) the modules43 and layers that make up the system and software, 2) the relationships among the modules and layers, 3) the data inputs/outputs and flow of data among the modules and layers, and 4) how users or external products, including IT infrastructure and peripherals (e.g., wirelessly connected medical devices) interact with the system and software. Sponsors should provide the appropriate level of detail in the system and software architecture diagram to convey the information in a manner that can facilitate an efficient premarket review, including descriptive text (in the diagram or in an accompanying document) to explain the architecture diagram where appropriate. A system and software architecture diagram that is not appropriately tailored (e.g., too high level, too detailed, or overly confusing) will likely increase requests for additional information from the FDA review staff. To the extent appropriate, the system and software architecture diagram can be communicated in one or more diagrams and in one or more formats, and may convey different dimensions of the system and software (e.g., cybersecurity architecture, state diagram). If more than one diagram is used, the sponsor should provide a high-level diagram that communicates the overview and points to the other diagrams that provide additional detail. The relationship between diagrams should also be clearly communicated. In general, the sponsor should take note of the following visual, language, and reference considerations when developing an effective system and software architecture diagram:
Visual Considerations
- The diagram and the means for communicating information in the diagram should be visually consistent (e.g., a solid arrow should convey a specific meaning as compared to a dashed arrow; icons should be used consistently; lines that intersect should clearly communicate whether the intersection is a crossing or connection) and the meaning ascribed should be clearly communicated (e.g., through the use of standard symbols and notation).
- The level of detail provided in the diagram should be consistent throughout unless areas of less detail are clearly explained (e.g., in the case of functions, within the system or software, that are not under review).
- Modules should be grouped in a logical and obvious manner.
- Use of color or other visual means (e.g., dashed boxes within solid boxes) should be used to convey layering within the system, software, or module.
- Visual clutter should be avoided, and the diagram should be scaled according to the complexity and amount of information presented.
Language Considerations
- Annotations should be used to provide additional information about a particular module or data element (e.g., a plain-language explanation of the module purpose or a pointer to a document or requirement within the premarket submission).
- Use of terminology and naming conventions should be consistent within the diagram and the remainder of the premarket submission materials.
- Use of acronyms, jargon, or terms that are not defined in the diagram itself should be avoided.
References
- The diagram should reference other documents in the submission (e.g., System & Software Description, Software Requirements Specification), as appropriate.
- For submissions related to modifications to a previously cleared or approved device, the diagram should identify and reference those modules that are affected by the modification.
The above considerations are intended to serve as a guide and may not apply in every case or may apply differently for different diagrams. When developing the system and software architecture diagram, manufacturers are encouraged to leverage industry best practices within and outside the medical device industry.
For multiple function device products, the system and software architecture diagram should clearly delineate between the device functions-under-review and the “other functions,” as that term is used in the guidance Multiple Function Device Product: Policy and Considerations. The system and software architecture diagram should include adequate detail to understand how or if the “other function(s)” interact with or impact the device function-under-review.
An example system and software architecture diagram is provided in Appendix B of this guidance, which illustrates one approach to effectively convey the recommended information to facilitate an efficient premarket review of a software system with multiple modules. The example demonstrates how the considerations described in this section can be implemented into a system and software architecture diagram. The modules in the example are intended for illustration purposes only and are not intended to document or represent a comprehensive or complete system and software architecture diagram for a specific medical device or system. The approach illustrated can be applied to any system and software architecture diagram, including standalone SaMD.
D. Risk Management File 🔗
The risk management file should be provided as part of the premarket submission and include the following documentation:
Risk Management Plan 🔗
- Refer to the FDA recognized consensus standard ISO 14971 for recommendations on the content of a risk management plan.
- Submit a risk management plan to support the effectiveness of the risk management activities and processes for a particular medical device. In FDA’s review of the risk management plan, the Agency intends to primarily focus on:
- Individual risk acceptability criteria including the need for risk reduction (control).
- Method to evaluate the acceptability of the overall residual risk for all residual risks after all risk control measures have been implemented and verified.
- The risk acceptability criteria should be based on the sponsor’s policy for determining acceptable risk. The acceptability criteria should be documented in the risk management plan before an initial risk evaluation is performed for the medical device software under review.
- It should be clear in the risk management plan how the manufacturer’s plans to evaluate the overall residual risk against the benefits of the intended use of the device. For SiMD, if this information is covered in the overall device hazard analysis, this should be noted in the software documentation and reference that section of the submission.
Risk Assessment 🔗
A risk assessment should be provided for all medical device software. For multiple function device products, the risk assessment should include the results of a risk based analysis of any potential adverse impact or labeled positive impact of “other function(s),” as that term is used in the guidance Multiple Function Device Product: Policy and Considerations, to the safety or effectiveness of the device function-under review.
The International Medical Device Regulators Forum (IMDRF) document, Software as a Medical Device: Possible Framework for Risk Categorization and Corresponding Considerations,44 may also be helpful in identifying probable risks. Specifically, it may be helpful to consider the significance of information provided by the software and the criticality of the medical situation or condition in which the software is intended to function.
All reasonably foreseeable software and hardware hazards associated with the device should be identified and documented in a tabular format, including those hazards resulting from intentional or reasonably foreseeable misuse of the device. You should consider the device’s intended use; the direct and indirect impacts to safety, treatment, and/or diagnosis; among other relevant considerations. In a tabular format, each identified hazard is captured in a singular line item which should include the following items:
- Identification of the hazard
- Cause(s) of the hazard
- Severity of the hazard
- Initial risk evaluation of hazard
- This includes assessment of acceptability (e.g., acceptable, not acceptable) and need for risk reduction (control) measures as defined in the risk management plan
- Risk Control Measures
- This should include the following risk control measures listed in descending order from highest to lowest priority:
- Design
- Protective measures (e.g., alarms)
- Information for safety (e.g., written warnings, on-screen warnings, training)
- There should be verification of the implementation of the risk control measures and verification of the effectiveness of the implemented risk control measures (i.e., the implemented risk control measure reduces risk). This can be accomplished by tracing the identified hazard to the verified specific risk control measures (e.g., a requirement ID in the SRS and SDS, a test name and identifier in the testing documentation that shows pass/fail results, a user manual name and identifier, a training manual name and identifier). For example, given an identified hazard (e.g., HAZ-XXX45), the following could be listed for traceability: a design related risk control measure that is documented in a software requirement specification (e.g., SRS-XXX) and software design specification (e.g., SDS-XXX), and tested as part of a unit test case (e.g., UT-XXX), integration test case (e.g., INT-XXX) and system test case (e.g., SYS-XXX). FDA recognizes that there may be instances where a specific hazard traces to several requirements/specifications and tests, thereby making the presentation of information cumbersome in the risk assessment document. Therefore, sponsors may choose to present this traceability in a separate table or document with line items to link together software requirements specifications, software design specifications, testing and identified hazards derived from the risk assessment.
- There should be an assessment of whether risk control measures introduce new hazards or hazardous situations or impact the initial risk evaluation.
- Document any risk control measures employed to mitigate increased risk or adverse effect on performance due to the combination of “other function(s),” as that term is used in the guidance Multiple Function Device Product: Policy and Considerations, with the device function under review.
- This should include the following risk control measures listed in descending order from highest to lowest priority:
- Residual risk evaluation after implemented risk control measures.
- This includes assessment of acceptability (e.g., acceptable, not acceptable) as defined in the risk management plan.
- Benefit-Risk Analysis
- If a residual risk is deemed not acceptable according to the acceptability criteria in the risk management plan and further risk control is not practicable, the manufacturer should provide documented evidence to demonstrate that the benefits of the intended use outweigh the residual risk.
Risk Management Report 🔗
- A risk management report should be provided to:
- Show how the risk management plan has been appropriately implemented.
- Demonstrate that the risk management file has been assessed by the appropriate personnel and the overall residual risk is acceptable
- Demonstrate appropriate methods are established for the collection and assessment of relevant production and post-production information
E. Software Requirements Specification (SRS) 🔗
The SRS documents the requirements46 for the software which typically specifies inputs and outputs, functions that the software will perform, hardware,47 programming language,48 compiler version, performance,49 interfaces,50 user interaction, error definition and handling, response times, intended operating environment, safety related requirements derived from a risk assessment (Refer to Section VI.D Risk Management File) and all ranges, limits, defaults, and specific values that the software will accept. For additional details on what should be included in the software requirements specification, refer to the guidance, General Principles of Software Validation.51
The QSR requires “a mechanism for addressing incomplete, ambiguous, or conflicting requirements.”52 Each requirement (e.g., hardware, software, user, operator interface, and safety) identified in the software requirements specification should be evaluated for accuracy, completeness, consistency, testability, correctness, and clarity.
A complete SRS document should be provided. The documentation should include a description of the software requirement identification and tracking methodology used to support the traceability of the requirements.
FDA acknowledges that modern development practices may employ incremental or evolutionary software development practices. Additional forms of software requirements might be included in the submission, such as well elaborated stories, use cases, textual descriptions, screen mockups, and control flows. Executable requirements may be provided with additional clarification to support readability.
In order to facilitate a timely premarket review, the following recommendations should be considered in preparing SRS documentation:
- Format the SRS to be well-organized, easily navigable and readable with the labeling and/or grouping of requirements by function.
- Note any relevant traceability between requirements listed in the SRS and information related to those requirements in other software documentation (e.g., SDS, System and Software Architecture Diagram, etc.).
- If the premarket submission involves a modification to an existing approved or cleared device, highlight all pertinent differences in software requirements.
- The manufacturer may choose to identify the requirements the manufacturer believes are most critical (i.e., could have the most significant impacts) to the device’s safety and effectiveness. These requirements can be highlighted within the complete SRS document and/or consolidated in a supplemental document that includes these requirements of interest in a summarized format. This technique will help facilitate the presentation of those requirements that most critically affect clinical functionality or performance specifications that are directly associated with the intended use of the device.
Documentation of requirements included in the premarket submission for the device function under-review should include adequate detail to describe any expected relationship, utility, reliance, or interoperability with any “other function,” as that term is used in the guidance Multiple Function Device Product: Policy and Considerations.
See Design Inputs: FAQs and Examples for practical advice and examples.
F. Software Design Specification (SDS) 🔗
The Software Design Specification (SDS) may contain both a high level summary of the design and detailed design information. In terms of the relationship between the Software Requirement Specification (SRS) and the SDS, the SRS describes what the software function will do and the SDS describes how the requirements in the SRS are implemented. The information presented in the SDS should be sufficient to ensure that the work performed by the software engineers who created the software device function was clear and unambiguous, with minimal ad hoc design decisions. Documentation of specifications included in the premarket submission for the device function-under-review should include adequate detail to describe any expected relationship, utility, reliance, or interoperability with any “other function,” as that term is used in the guidance Multiple Function Device Product: Policy and Considerations.
(1) Basic Documentation Level
No SDS documentation recommended in the premarket submission.
(2) Enhanced Documentation Level
A singular SDS document or set of SDS documents that provide the technical design details of how the software functions, how the software design completely and correctly implements all the requirements of the SRS and how the software design traces to the SRS in terms of intended use, functionality, safety, and effectiveness. The software functional units or modules along with the interfaces among them identified in the architectural (i.e., high level) design should be documented with the corresponding detailed (i.e., low-level) design information in the SDS. The information provided for review should be sufficient to ensure that the work performed in developing the software functional units or modules and their interfaces was clear and unambiguous, with minimal ad hoc design decisions. For example, the creation of the SDS is expected to have occurred as a prospective activity where the SDS was used to guide the design, development and testing of the software rather than documented retrospectively after the software design has been implemented by ad hoc design methods.
For additional details on what should be included in the software design specification, refer to the guidance, General Principles of Software Validation.53
G. Software Development and Maintenance Practices 🔗
(1) Basic Documentation Level
For devices that the Basic Documentation Level is appropriate, a Declaration of Conformity to the currently FDA-recognized version of ANSI/AAMI IEC 62304 Medical Device Software - Software Life Cycle Processes may suffice.
Alternatively, a summary of the processes and procedures that are in place to manage the software life cycle development, software configuration and change management and software maintenance activities could be provided. This summary information should include an adequate description of:
- Processes and procedures used in software development, verification and validation.
- Standards (e.g., coding), methods and tools used in software development.
- Main deliverables of the typical activities and tasks involved in software development, verification and validation.
- Processes, procedures, and tools used to link user needs, system requirements, software requirements, software design specifications, software testing and implemented risk control measures (i.e., traceability).
- Processes and procedures used in software configuration and change management.
- Processes and procedures used in software maintenance that includes risk assessment of software changes, initial testing that evaluates the correctness of the implemented software change(s) and regression analysis and testing.
- Note: Software maintenance typically includes the following types of software changes:
- Corrective: Changes made to correct errors and faults in the software
- Perfective: Changes made to the software to improve the performance, maintainability, or other attributes of the software system
- Adaptive: Changes to make the software system usable in a changed environment
- Note: Software maintenance typically includes the following types of software changes:
(2) Enhanced Documentation Level
For devices that the Enhanced Documentation Level is appropriate, a Declaration of Conformity to the currently FDA-recognized version of ANSI/AAMI IEC 62304 Medical Device Software - Software Life Cycle Processes may suffice.
Alternatively, a complete configuration management and maintenance plan document could be provided in addition to the summary documentation requested for the Basic Documentation Level, as described above.
H. Software Testing as part of Verification and Validation 🔗
Refer to Section IV (Definitions) for important information pertaining to FDA’s thinking on verification and validation, as it relates to this guidance. Additionally, please refer to guidance General Principles of Software Validation54 for additional details regarding FDA’s thinking regarding software testing particularly unit level (module or component) testing, integration level (internal and external interfaces) testing and system level (functional) testing.
Software testing may sometimes involve other device performance testing activities. If the premarket submission leverages information from the performance testing section to address software validation content, the manufacturer is encouraged to appropriately reference the performance testing material to facilitate the navigation between submission sections, reduce instances of duplication, and improve readability.
(1) Basic Documentation Level
The following software testing documentation should be provided:
- A summary description of the testing activities at the unit, integration, and system levels. The summary description should include the software version tested and the overall pass/fail test results for all test protocols (i.e., collection of test procedures for specific software functionality) executed. If the device is a modified version of a previously cleared or approved device, provide a summary of the modifications compared with the previous cleared or approved version.
- Any intentional changes made in response to failed tests and documentation of test results demonstrating that the intentional changes were implemented correctly.
- A regression analysis and pass/fail test results to account for unintended effects of a software change.
- Regression analysis is the determination of the impact of a change based on review of the relevant documentation (e.g., software requirements specification, software design specification, source code, test plans, test cases, test scripts, etc.) in order to identify the necessary regression tests to be run. Regression testing is the rerunning of test cases that a program has previously executed correctly and comparing the current result to the previous result in order to detect unintended effects of a software change.
- System level test protocol including expected results derived from software requirements, actual results that are observed and recorded, objective pass/fail determination (i.e., actual results are acceptably equivalent to expected results) and a system level test report. The system level test report should demonstrate that the protocol has been acceptably executed with passing test results and any unresolved anomalies have been acceptably deferred based on a risk assessment for the candidate release version.
(2) Enhanced Documentation Level
In addition to the documentation requested for the Basic Documentation Level, unit and integration level test protocols and reports should be provided, including expected results derived from software requirements and design, actual results that are observed and recorded, objective pass/fail determination (i.e., actual results are acceptably equivalent to expected results) and unit and integration test reports. The unit and integration level test reports should demonstrate that the protocols have been acceptably executed with passing testing results and any unresolved anomalies have been acceptably deferred based on a risk assessment for the candidate release version.
I. Revision Level History 🔗
The documentation should include the history of software revisions generated during product development.
This typically takes the form of a line-item tabulation of the major changes to the software during the development cycle, including date, version number, a brief description of the changes in the version relative to the previous version, and an indication of the version testing was performed on, including bench testing, animal testing, and clinical testing, if applicable.
The last entry in the list should be the final version to be incorporated in the released device. This entry should also include any differences between the tested version of software and the released version, along with an assessment of the potential effect of the differences on the safety and effectiveness of the device.
If a manufacturer’s development practices use an iterative methodology, information on any changed software requirements and how they continue to meet the system requirements or design inputs should be provided.
If the revision level history includes a version(s) that corresponds to a previously released version of the software, please highlight in the revision level history document each prior released version and (if applicable) the premarket submission number(s) associated with that release.
J. Unresolved Anomalies (e.g., Bugs, Defects, or Errors) 🔗
An anomaly is any condition that deviates from the expected behavior based on requirements specifications, design documents, standards, or from someone’s perceptions or experiences.
For each unresolved anomaly, indicate the:
- problem;
- impact on device performance; and
- any plans or timeframes for correcting the problem (where appropriate).
Each item should be annotated with an explanation of the impact of the anomaly on device safety or effectiveness, including operator usage and human factors issues. Typically, this anomaly list can be generated as an output of a change control board or similar mechanism for evaluation and disposition of unresolved software anomalies. FDA suggests ranking anomalies based on risk to patient and/or operator (user). Additionally, the Agency recommends including defect classification for each anomaly using a defect taxonomy, such as ANSI/AAMI SW91’s Classification of defects in health software.
If the resolution of any unresolved anomalies will be deferred, a risk-based rationale for why each unresolved anomaly would not impact device safety or effectiveness should be provided.
A list of unresolved anomalies should be communicated to end user(s) as appropriate to assist in the proper operation of the device. In all instances where it is practical to do so, any mitigations or possible work-arounds for unresolved anomalies should be included in the premarket submission.
VII. Additional Information 🔗
A. Regulatory Considerations for Software Functions
Section 3060(a) of the Cures Act amended section 520 of the FD&C Act on December 13, 2016, removing certain software functions from the definition of device in section 201(h) of the FD&C Act. To learn more about FDA’s regulatory considerations for software functions, please consider the following reference materials:
- Changes to Existing Medical Software Policies Resulting from Section 3060 of the 21st Century Cures Act55
- General Wellness: Policy for Low Risk Devices56
- Policy for Device Software Functions and Mobile Medical Applications57
- Medical Device Data Systems, Medical Image Storage Devices, and Medical Image Communications Devices58
- How to Determine if Your Product is a Medical Device59
B. Off-The-Shelf (OTS) Software Use in Medical Devices
FDA recognizes the Off-The-Shelf Software Use in Medical Devices (“OTS”) guidance uses “Level of Concern” factors to determine the software content for OTS software. Therefore, after finalization of this guidance, FDA intends to update the OTS guidance to be consistent with this guidance. For example, Table 2 below is derived from “Table 1” in the OTS guidance. Table 2 incorporates the Documentation Levels described in this guidance to help explain how the concepts in this guidance could be represented in a future update to the OTS guidance
Table 2: Documentation Level for Off-The-Shelf Software
| Basic Documentation Level | Enhanced Documentation Level |
|---|---|
| Hazard Analysis | Hazard Analysis |
| Basic Documentation | Basic Documentation |
| Hazard Mitigations | Hazard Mitigations |
| Describe and Justify Residual Risk | Describe and Justify Residual Risk |
| Special Documentation |
<table>
<tr>
<th>Basic Documentation Level</th>
<th>Enhanced Documentation Level</th>
</tr>
<tr>
<td>Hazard Analysis</td>
<td>Hazard Analysis</td>
</tr>
<tr>
<td>Basic Documentation</td>
<td>Basic Documentation</td>
</tr>
<tr>
<td>Hazard Mitigations</td>
<td>Hazard Mitigations</td>
</tr>
<tr>
<td>Describe and Justify Residual Risk</td>
<td>Describe and Justify Residual Risk</td>
</tr>
<tr>
<td></td>
<td>Special Documentation</td>
</tr>
</table>
Please refer to the OTS guidance for additional information on what information is requested as
part of “hazard analysis,” “basic documentation,”60 “hazard mitigations,” “describe and justify residual risk,” and “special documentation” content. The OTS Guidance describes “Special Documentation” in section V.E. The information requested as part of the “Special Documentation” includes: (1) an assurance to FDA that the product development methodologies used by the OTS software developer are appropriate and sufficient for the intended use of the OTS software within the specific medical device, (2) the procedures and results of the verification and validation activities performed for the OTS software are appropriate and sufficient for the safety and effectiveness requirements of the medical device, and (3) the existence of appropriate mechanisms for assuring the continued maintenance and support of the OTS software should the original OTS software developer terminate their support.
When appropriate, the documentation described in this guidance can be leveraged to address the content described in the OTS guidance. For example, the OTS Hazard Analysis information may be captured as part of the Risk Assessment, as described in this guidance.
Please note, OTS software is typically an “other function,” as that term is used in the guidance Multiple Function Device Product: Policy and Considerations. Therefore, please consider the impact of the OTS software on the device function-under-review and provide information (as recommend in the OTS guidance) about the impact on the device function from the OTS software in a manner consistent with the policy described in the Multiple Function Device Product: Policy and Considerations guidance.
Additionally, please note the term SOUP, Software of Unknown Pedigree, may also be used to describe software from a third party. For additional information on the term SOUP, please refer to the use of the term in the standard ANSI/AAMI/IEC 62304: Medical Device Software - Software Life Cycle Processes.
C. Comparison of Guidance to IEC 62304 and 840 ANSI/AAMI/IEC 62304
FDA recognizes voluntary consensus standards to help facilitate meeting requirements under the statute or regulations. The use of FDA-recognized consensus standards can increase predictability, streamline premarket review, provide clearer regulatory expectations, facilitate market entry for safe and effective medical products, and promote international harmonization (see Recognition and Withdrawal of Voluntary Consensus Standards61).
As of this draft guidance’s publication, both IEC 62304 Edition 1.1 and ANSI/AAMI/IEC
848 62304:2006/A1:2016 Medical device software — Software life cycle processes (henceforth “IEC 62304”), are FDA-recognized voluntary consensus standards. The scope of each standard includes life cycle requirements for medical device software, including the set of processes, activities, and tasks that establish a common framework for medical device software life cycle processes.
There are similarities and differences between the intents and information discussed in this guidance and IEC 62304. Both this guidance and IEC 62304 discuss how to document and communicate software development, maintenance, and risk management processes. However, this guidance document focuses on documentation and communication only as it pertains to facilitating FDA’s complete and efficient review of device software functions subject to premarket submission, whereas the scope of IEC 62304 extends beyond the intent of this guidance and discusses broader perspectives that may not result in documents that are easily reviewed by FDA.
Therefore, this guidance document intends to leverage IEC 62304 in a very specific manner in order to reduce the amount of documentation requested to support a premarket submission for a device software function. Specifically, Section V.G: Software Development and Maintenance Practices offers an alternate approach through which FDA intends to accept a Declaration of Conformity to IEC 62304 in place of a detailed description of software development and maintenance practices (formerly, “Software Development Environment Description”). Sponsors may refer to the guidance document Appropriate Use of Voluntary Consensus Standards inmarket Submissions for Medical Devices62 for more information on preparation of a Declaration of Conformity. Conformity to this consensus standard is voluntary and, as such, an alternate approach is offered. When assessing the appropriate Documentation Level for the device and the overall recommended documentation for inclusion in a premarket submission, please refer to Section V (Documentation Level) and Section VI (Recommended Documentation) of this guidance.
Appendix A: Documentation Level Examples 🔗
The following are examples of devices that demonstrate the implementation of the Documentation Level factors. Please note that these generalized examples do not necessarily account for every possible detail, risk, or consideration a sponsor should evaluate, and should not be taken to mean that the devices described definitely do or do not require a certain Documentation Level. These examples do not define the appropriate Documentation Level for a particular device type. As such, the Documentation Level should be uniquely considered for each particular device or device modification and in consideration of the device’s intended use. When addressing the recommendations in sections V and VI.A of this guidance, FDA encourages sponsors to leverage their device’s risk assessment when providing a rationale for choosing a Documentation Level.
-
A non-contact infrared thermometer intended for intermittent measurement of body temperature from the forehead
# Factors Yes/No 1 The device is a constituent part of a combination product. No 2 The device (a) is intended to test blood donations for transfusion-transmitted infections; or (b) is used to determine donor and recipient compatibility; or (c) is Blood Establishment Computer Software. No 3 The device is classified as class III. No 4 A failure or latent flaw of the device software function(s) could present a probable risk of death or serious injury either to the patient, user of the device, or others in the environment of use. These risk(s) should be assessed prior to implementation of risk control measures. You should consider the risk(s) in the context of the device’s intended use; the direct and indirect impacts to safety, treatment, and/or diagnosis; and other relevant considerations. No <table> <tr> <th>#</th> <th>Factors</th> <th>Yes/No</th> </tr> <tr> <td>1</td> <td>The device is a constituent part of a combination product.</td> <td>No</td> </tr> <tr> <td>2</td> <td>The device (a) is intended to test blood donations for transfusion-transmitted infections; or (b) is used to determine donor and recipient compatibility; or (c) is Blood Establishment Computer Software.</td> <td>No</td> </tr> <tr> <td>3</td> <td>The device is classified as class III.</td> <td>No</td> </tr> <tr> <td>4</td> <td>A failure or latent flaw of the device software function(s) could present a probable risk of death or serious injury either to the patient, user of the device, or others in the environment of use. These risk(s) should be assessed prior to implementation of risk control measures. You should consider the risk(s) in the context of the device’s intended use; the direct and indirect impacts to safety, treatment, and/or diagnosis; and other relevant considerations.</td> <td>No</td> </tr> </table>Outcome: The device is class II and is not a constituent part of a combination product, not Blood Establishment Computer Software, and is not intended for use in testing blood donations for transfusion-transmitted infections or determining donor and recipient compatibility. A failure or latent flaw of the device software function(s) would not present a probable risk of death or serious injury to either a patient, user of the device, or others in the environment of use prior to the implementation of risk control measures. Therefore, this device would fall under “Basic Documentation Level.”
-
An implantable cardiac pacemaker used to treat bradycardia.
# Factors Yes/No 1 The device is a constituent part of a combination product. No 2 The device (a) is intended to test blood donations for transfusion-transmitted infections; or (b) is used to determine donor and recipient compatibility; or (c) is Blood Establishment Computer Software. No 3 The device is classified as class III. Yes 4 A failure or latent flaw of the device software function(s) could present a probable risk of death or serious injury either to the patient, user of the device, or others in the environment of use. These risk(s) should be assessed prior to implementation of risk control measures. You should consider the risk(s) in the context of the device’s intended use; the direct and indirect impacts to safety, treatment, and/or diagnosis; and other relevant considerations. Yes <table> <tr> <th>#</th> <th>Factors</th> <th>Yes/No</th> </tr> <tr> <td>1</td> <td>The device is a constituent part of a combination product.</td> <td>No</td> </tr> <tr> <td>2</td> <td>The device (a) is intended to test blood donations for transfusion-transmitted infections; or (b) is used to determine donor and recipient compatibility; or (c) is Blood Establishment Computer Software.</td> <td>No</td> </tr> <tr> <td>3</td> <td>The device is classified as class III.</td> <td>Yes</td> </tr> <tr> <td>4</td> <td>A failure or latent flaw of the device software function(s) could present a probable risk of death or serious injury either to the patient, user of the device, or others in the environment of use. These risk(s) should be assessed prior to implementation of risk control measures. You should consider the risk(s) in the context of the device’s intended use; the direct and indirect impacts to safety, treatment, and/or diagnosis; and other relevant considerations.</td> <td>Yes</td> </tr> </table>Outcome: The device is not a constituent part of a combination product, is not Blood Establishment Computer Software, and is not intended for use in testing blood donations for transfusion-transmitted infections or determining donor and recipient compatibility. However, the device is classified as a class III device. Furthermore, a failure or latent flaw of the device software function(s) (e.g., algorithm operates as designed but incorrectly senses an ectopic beat) would present a probable risk of death or serious injury to the patient prior to the implementation of risk control measures. Therefore, this device would fall under “Enhanced Documentation Level.”
-
A facility use continuous ventilator
# Factors Yes/No 1 The device is a constituent part of a combination product. No 2 The device (a) is intended to test blood donations for transfusion-transmitted infections; or (b) is used to determine donor and recipient compatibility; or (c) is Blood Establishment Computer Software. No 3 The device is classified as class III. No 4 A failure or latent flaw of the device software function(s) could present a probable risk of death or serious injury either to the patient, user of the device, or others in the environment of use. These risk(s) should be assessed prior to implementation of risk control measures. You should consider the risk(s) in the context of the device’s intended use; the direct and indirect impacts to safety, treatment, and/or diagnosis; and other relevant considerations. Yes <table> <tr> <th>#</th> <th>Factors</th> <th>Yes/No</th> </tr> <tr> <td>1</td> <td>The device is a constituent part of a combination product.</td> <td>No</td> </tr> <tr> <td>2</td> <td>The device (a) is intended to test blood donations for transfusion-transmitted infections; or (b) is used to determine donor and recipient compatibility; or (c) is Blood Establishment Computer Software.</td> <td>No</td> </tr> <tr> <td>3</td> <td>The device is classified as class III.</td> <td>No</td> </tr> <tr> <td>4</td> <td>A failure or latent flaw of the device software function(s) could present a probable risk of death or serious injury either to the patient, user of the device, or others in the environment of use. These risk(s) should be assessed prior to implementation of risk control measures. You should consider the risk(s) in the context of the device’s intended use; the direct and indirect impacts to safety, treatment, and/or diagnosis; and other relevant considerations.</td> <td>Yes</td> </tr> </table>Outcome: The device is not a constituent part of a combination product, is not Blood Establishment Computer Software, and is not intended for use in testing blood donations for transfusion-transmitted infections or determining donor and recipient compatibility. The device is class II. However, a failure or latent flaw of the device software function(s) (e.g., exploited cybersecurity vulnerability compromises device functionality) would present a probable risk of death or serious injury to a patient (e.g., loss of life supporting function) prior to the implementation of risk control measures. Therefore, this device would fall under “Enhanced Documentation Level.”
-
A Blood Establishment Computer Software or BECS accessory
# Factors Yes/No 1 The device is a constituent part of a combination product. No 2 The device (a) is intended to test blood donations for transfusion-transmitted infections; or (b) is used to determine donor and recipient compatibility; or (c) is Blood Establishment Computer Software. Yes 3 The device is classified as class III. No 4 A failure or latent flaw of the device software function(s) could present a probable risk of death or serious injury either to the patient, user of the device, or others in the environment of use. These risk(s) should be assessed prior to implementation of risk control measures. You should consider the risk(s) in the context of the device’s intended use; the direct and indirect impacts to safety, treatment, and/or diagnosis; and other relevant considerations. Yes <table> <tr> <th>#</th> <th>Factors</th> <th>Yes/No</th> </tr> <tr> <td>1</td> <td>The device is a constituent part of a combination product.</td> <td>No</td> </tr> <tr> <td>2</td> <td>The device (a) is intended to test blood donations for transfusion-transmitted infections; or (b) is used to determine donor and recipient compatibility; or (c) is Blood Establishment Computer Software.</td> <td>Yes</td> </tr> <tr> <td>3</td> <td>The device is classified as class III.</td> <td>No</td> </tr> <tr> <td>4</td> <td>A failure or latent flaw of the device software function(s) could present a probable risk of death or serious injury either to the patient, user of the device, or others in the environment of use. These risk(s) should be assessed prior to implementation of risk control measures. You should consider the risk(s) in the context of the device’s intended use; the direct and indirect impacts to safety, treatment, and/or diagnosis; and other relevant considerations.</td> <td>Yes</td> </tr> </table>Outcome: The device is not a constituent part of a combination product. It is Blood Establishment Computer Software. The device is class II. However, a failure or latent flaw of the device software function(s) (e.g., exploited cybersecurity vulnerability compromises device functionality) would present a probable risk of death or serious injury to a patient (e.g., loss of life supporting function) prior to the implementation of risk control measures. Therefore, this device would fall under “Enhanced Documentation Level.”
-
A qualitative in vitro nucleic acid screening test for the direct detection of Babesia DNA and RNA in whole blood samples from individual human donors.
# Factors Yes/No 1 The device is a constituent part of a combination product. No 2 The device (a) is intended to test blood donations for transfusion-transmitted infections; or (b) is used to determine donor and recipient compatibility; or (c) is Blood Establishment Computer Software. Yes 3 The device is classified as class III. No 4 A failure or latent flaw of the device software function(s) could present a probable risk of death or serious injury either to the patient, user of the device, or others in the environment of use. These risk(s) should be assessed prior to implementation of risk control measures. You should consider the risk(s) in the context of the device’s intended use; the direct and indirect impacts to safety, treatment, and/or diagnosis; and other relevant considerations. Yes <table> <tr> <th>#</th> <th>Factors</th> <th>Yes/No</th> </tr> <tr> <td>1</td> <td>The device is a constituent part of a combination product.</td> <td>No</td> </tr> <tr> <td>2</td> <td>The device (a) is intended to test blood donations for transfusion-transmitted infections; or (b) is used to determine donor and recipient compatibility; or (c) is Blood Establishment Computer Software.</td> <td>Yes</td> </tr> <tr> <td>3</td> <td>The device is classified as class III.</td> <td>No</td> </tr> <tr> <td>4</td> <td>A failure or latent flaw of the device software function(s) could present a probable risk of death or serious injury either to the patient, user of the device, or others in the environment of use. These risk(s) should be assessed prior to implementation of risk control measures. You should consider the risk(s) in the context of the device’s intended use; the direct and indirect impacts to safety, treatment, and/or diagnosis; and other relevant considerations.</td> <td>Yes</td> </tr> </table>Outcome: The device is not a constituent part of a combination product and is not Blood Establishment Computer Software. The device is licensed under a BLA and consequently is not class III. However, the device is intended for use in testing blood donations for a transfusion-transmitted infection where an inaccurate result could result in blood recipient death or serious injury prior to the implementation of risk control measures. Therefore, this device would fall under “Enhanced Documentation Level.”
-
A hardware-only hip prosthesis. Outcome: This device would not have a Documentation Level because it does not contain software.
Appendix B: System and Software Architecture Diagram Chart Examples 🔗
The examples are intended for illustration purposes only and are not intended to document a comprehensive system and software architecture diagram for a specific medical device or system. The approach illustrated can be applied to any system and software architecture diagram, including standalone SaMD. The examples demonstrate how the considerations described in section VI.C (System and Software Architecture Design Chart) can be implemented into a system and architecture diagram. The example is not intended to represent a complete system and architecture diagram.
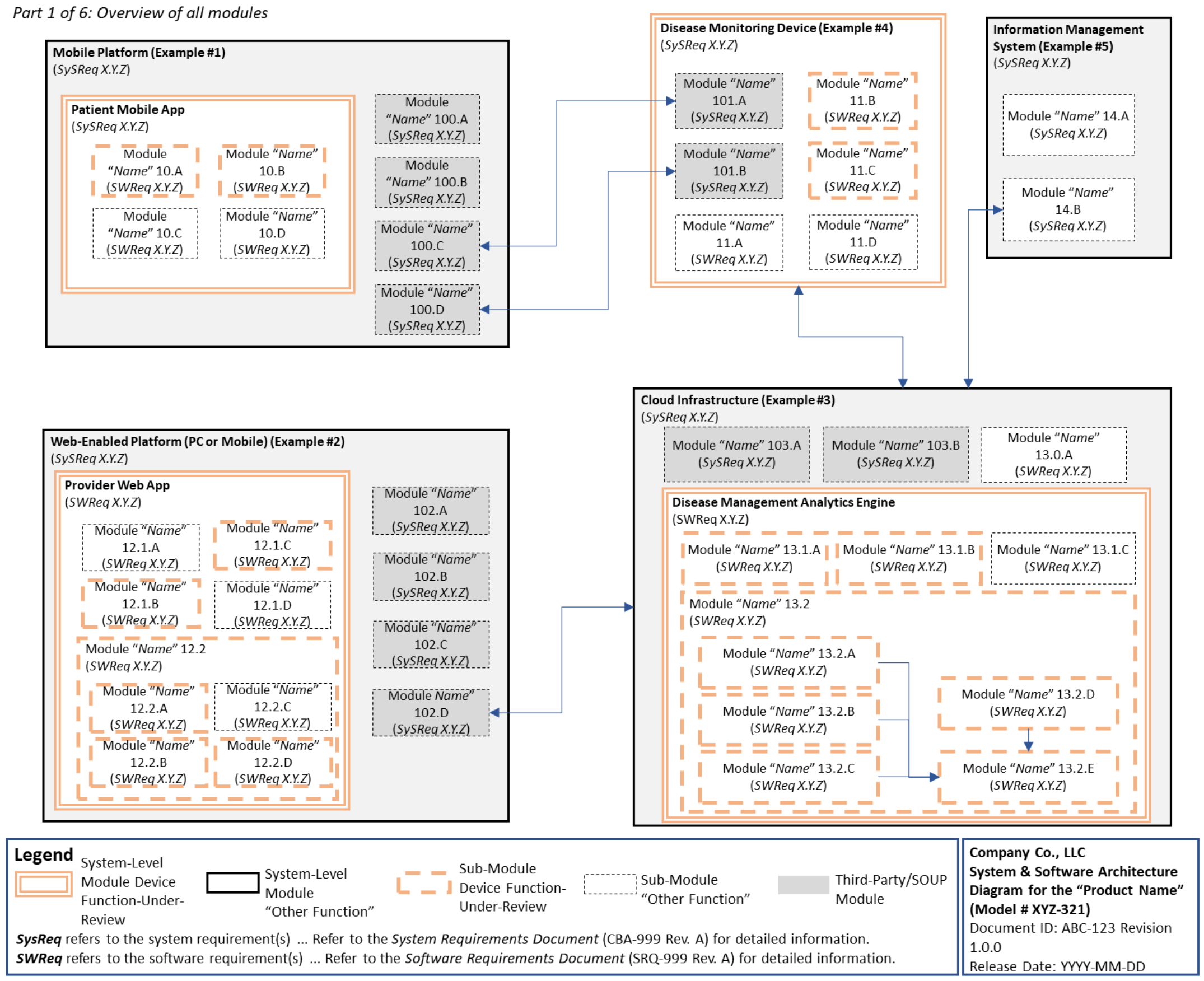
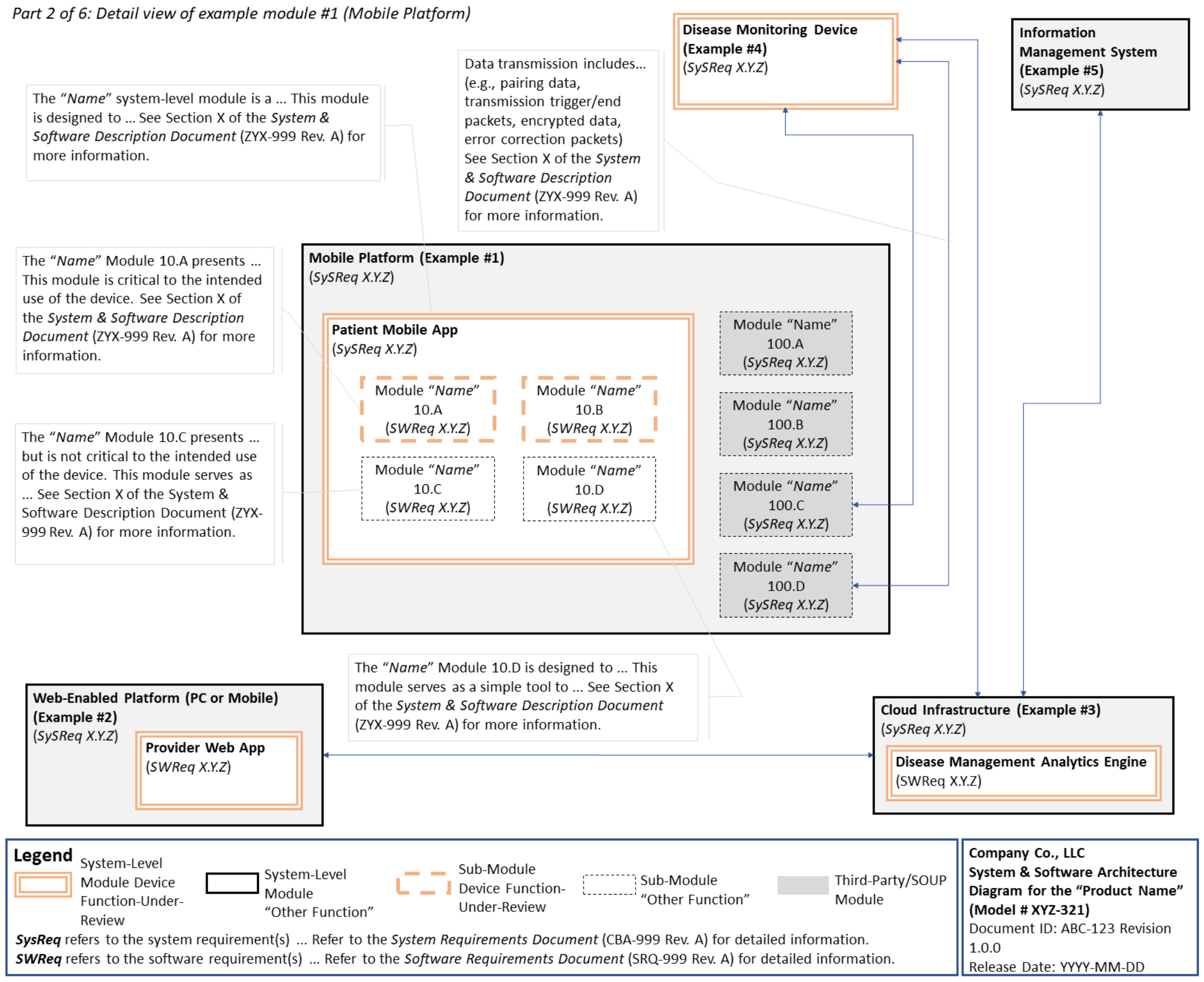
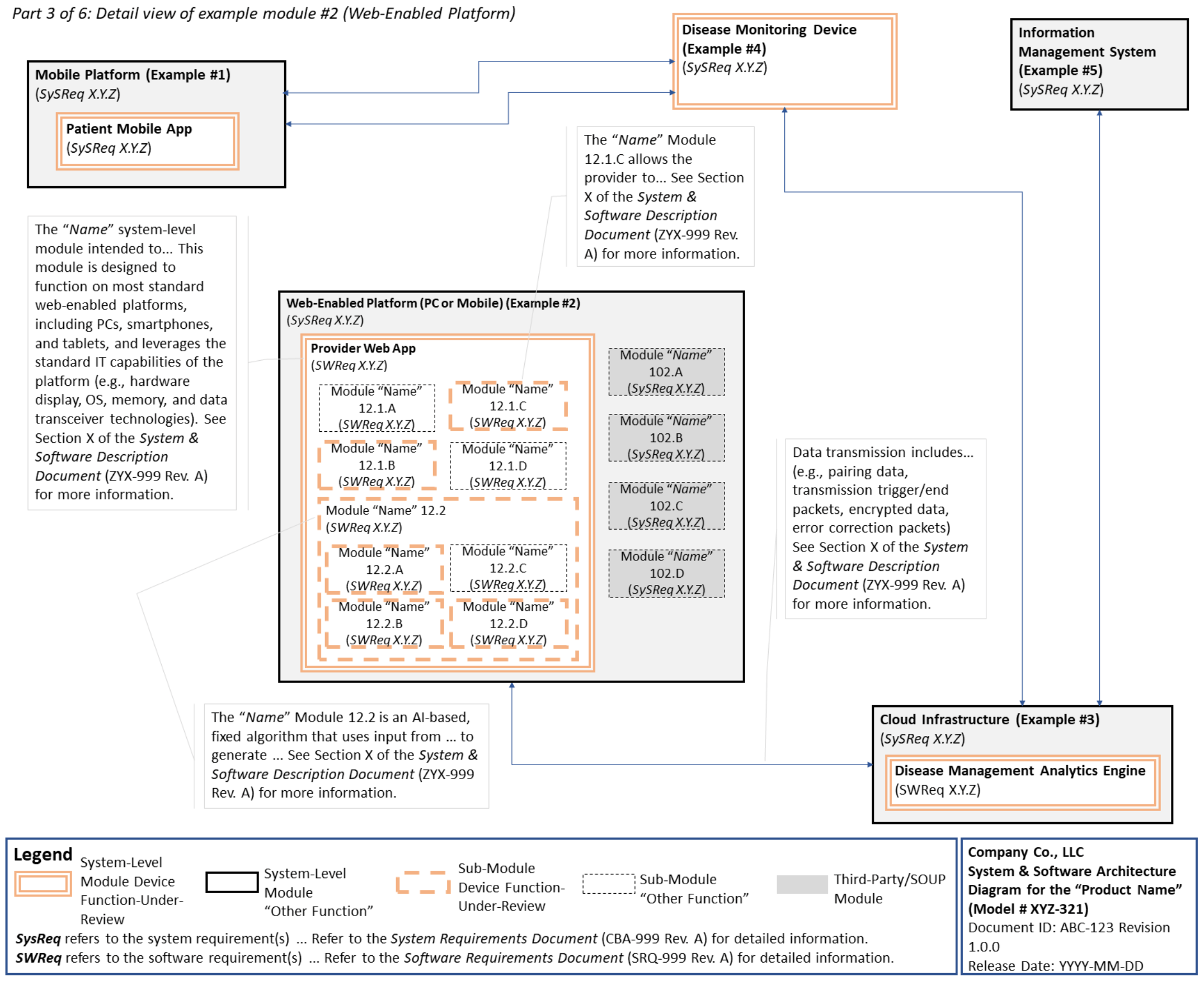
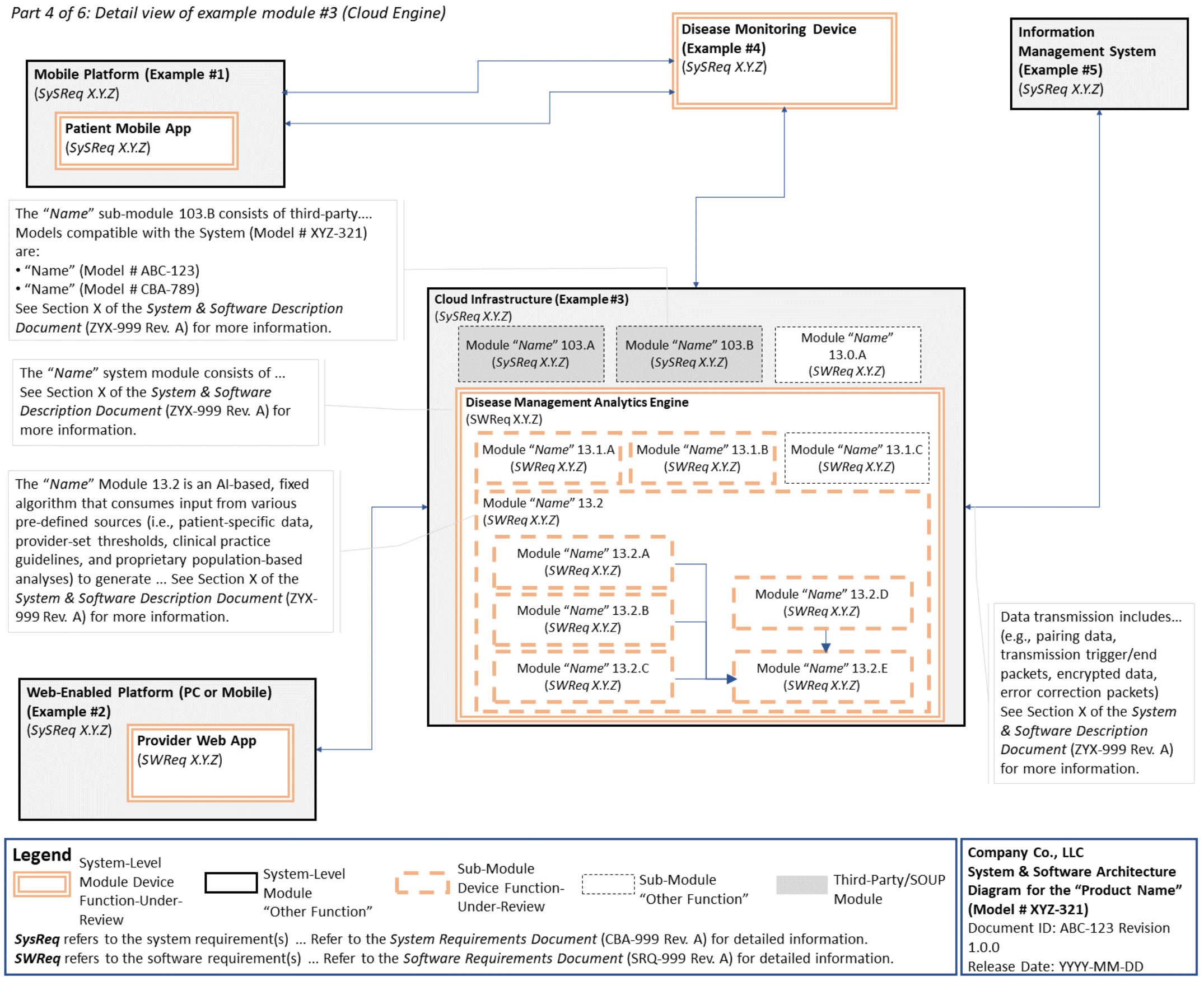
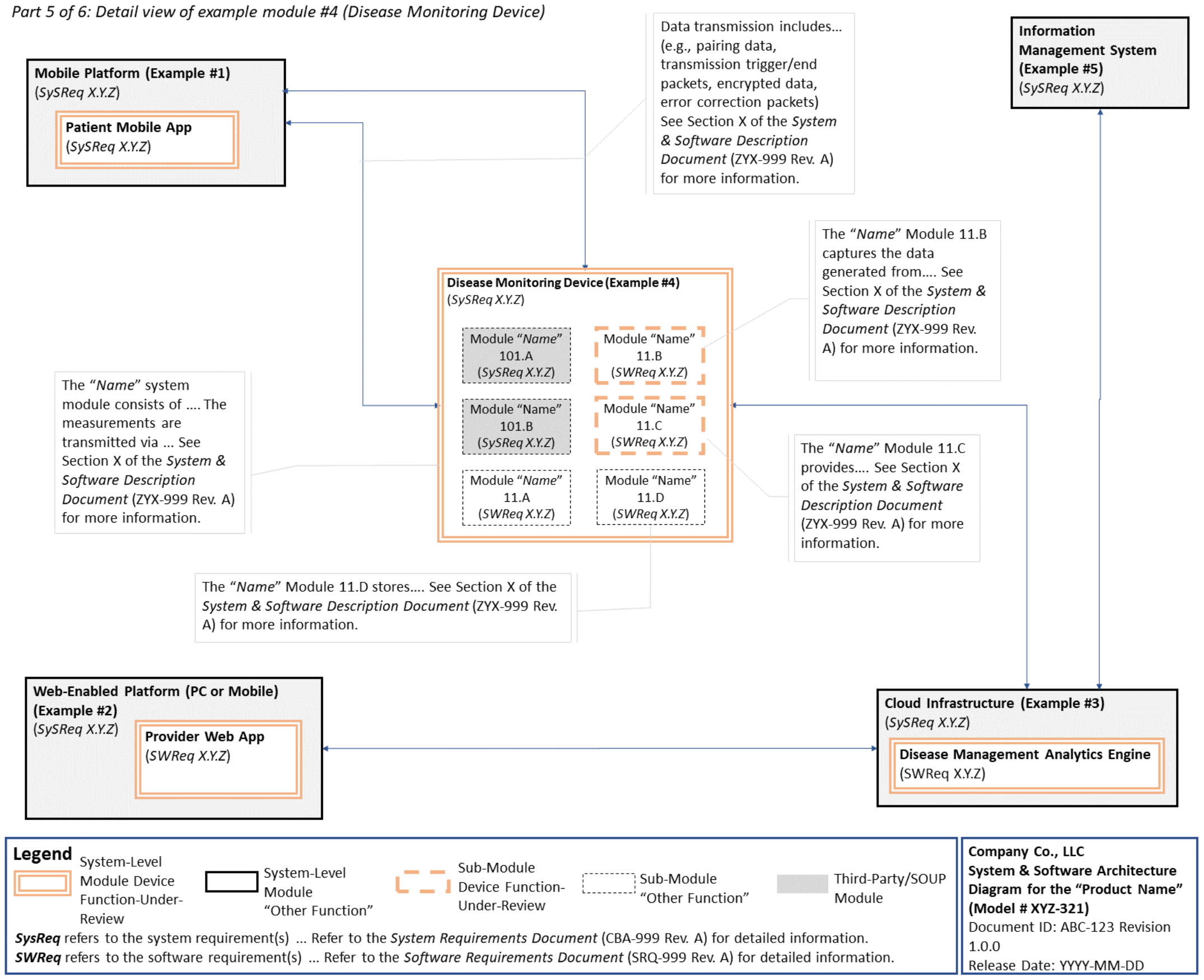
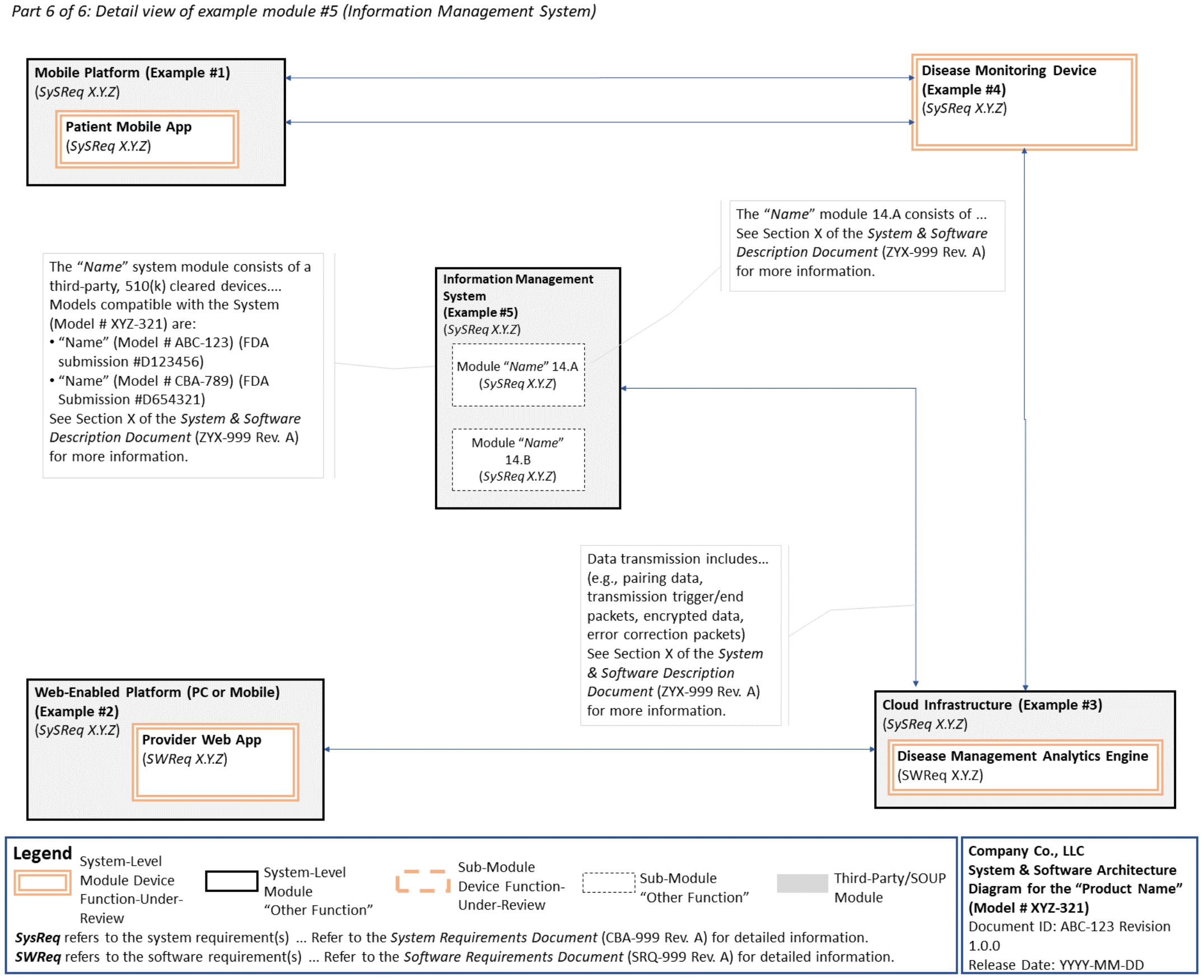
We can help 🔗
If you're overwhelmed by cybersecurity requirements for your SaMD, we can help. We provide turn-key engineering solutions and regulatory consulting services. Contact us if you'd like a team that help bring your device to the market as quickly as possible. If you have an engineering team, we can also integrate cybersecurity into your quality system.
Footnotes 🔗
-
The term “device” is defined in 201(h) of the Federal Food, Drug, and Cosmetic (FD&C) Act to include an “instrument, apparatus, implement, machine, contrivance, implant, in vitro reagent, or other similar or related article, including any component, part, or accessory, which is ...intended for use in the diagnosis of disease or other conditions, or in the cure, mitigation, treatment, or prevention of disease, in man ... or intended to affect the structure or any function of the body of man...” and “does not include software functions excluded pursuant to section 520(o) of the FD&C Act.” ↩
-
See FDA website on “Software as a Medical Device (SaMD),” available at https://www.fda.gov/medicaldevices/digital-health-center-excellence/software-medical-device-samd. ↩
-
Available at https://www.fda.gov/regulatory-information/search-fda-guidance-documents/guidance-contentpremarket-submissions-software-contained-medical-devices. ↩
-
As a reminder, manufacturers of device software must create and maintain software-related documentation in accordance with the requirements of the Quality System (QS) Regulation (21 CFR 820.30 Subpart C – Design Controls of the Quality System Regulation). ↩
-
Available at https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfStandards/search.cfm. ↩
-
Available at https://www.fda.gov/regulatory-information/search-fda-guidance-documents/appropriate-usevoluntary-consensus-standards-premarket-submissions-medical-devices. ↩
-
Available at https://www.fda.gov/regulatory-information/search-fda-guidance-documents/standards-developmentand-use-standards-regulatory-submissions-reviewed-center-biologics-evaluation. ↩
-
Available at https://www.fda.gov/medical-devices/digital-health-center-excellence/guidances-digital-healthcontent. ↩
-
Available at https://www.fda.gov/regulatory-information/search-fda-guidance-documents/multiple-functiondevice-products-policy-and-considerations. ↩
-
Available at https://www.fda.gov/regulatory-information/search-fda-guidance-documents/shelf-software-usemedical-devices. ↩
-
See section VII.B to learn more about referencing Off-The-Shelf (OTS) Software Use in Medical Devices. ↩
-
Available at https://www.fda.gov/regulatory-information/search-fda-guidance-documents/design-considerationsand-premarket-submission-recommendations-interoperable-medical-devices. ↩
-
Available at https://www.fda.gov/regulatory-information/search-fda-guidance-documents/general-principlessoftware-validation. ↩
-
Available at https://www.fda.gov/regulatory-information/search-fda-guidance-documents/content-premarketsubmissions-management-cybersecurity-medical-devices-0. ↩
-
Available at https://www.fda.gov/regulatory-information/search-fda-guidance-documents/cybersecuritynetworked-medical-devices-containing-shelf-ots-software. ↩
-
For the purposes of this guidance, a software function that is “part of a hardware device” is software used to control a hardware device, or software necessary for a hardware device to achieve its intended use. ↩
-
Ibid ↩
-
Additional resources on determining whether a software product contains device functions are provided in section VII.A. ↩
-
As part of Quality System Regulation production and process controls, 21 CFR 820.70(i) states, “When computers or automated data processing systems are used as part of production or the quality system, the manufacturer shall validate computer software for its intended use according to an established protocol. All software changes shall be validated before approval and issuance. These validation activities and results shall be documented.” ↩
-
21 CFR 3.2(e). ↩
-
Section 201(h) of the FD&C Act. ↩
-
As part of the software validation and risk analysis required by 21 CFR 820.30(g), software device manufacturers may need to establish a cybersecurity vulnerability and management approach, where appropriate. FDA recommends that this approach include a set of cybersecurity design controls to ensure medical device cybersecurity and maintain medical device safety and effectiveness. Such design controls may make it more likely that FDA will find your device meets its applicable statutory standard for premarket review. ↩
-
Available at https://www.fda.gov/regulatory-information/search-fda-guidance-documents/general-principlessoftware-validation. ↩
-
See footnotes 1 and 2. ↩
-
Available at https://www.fda.gov/regulatory-information/search-fda-guidance-documents/shelf-software-usemedical-devices. ↩
-
See section VII.B to learn more about referencing Off-The-Shelf Software Use in Medical Devices. ↩
-
x as defined in 21 CFR 803.3(w). ↩
-
For examples, refer to the following FDA webpage: https://www.fda.gov/medical-devices/software-medicaldevice-samd/what-are-examples-software-medical-device. ↩
-
See footnotes 1 and 2. ↩
-
See footnote 16. ↩
-
See footnotes 1 and 2. ↩
-
See footnote 16. ↩
-
See “General Principles of Software Validation” Guidance, available at https://www.fda.gov/regulatoryinformation/search-fda-guidance-documents/general-principles-software-validation. ↩
-
See 21 CFR 801.4 (“…[I]ntended uses… refer to the objective intent of the persons legally responsible for the labeling of an article. The intent may be shown by such persons’ expressions, the design or composition of the article, or by the circumstances surrounding the distribution of the article.”) ↩
-
21 CFR 3.2(e). ↩
-
Blood establishment computer software (BECS) is a device used in the manufacture of blood and blood components to assist in the prevention of disease in humans by identifying ineligible donors, by preventing the release of unsuitable blood and blood components for transfusion or for further manufacturing into products for human treatment or diagnosis, by performing compatibility testing between donor and recipient, or by performing positive identification of patients and blood components at the point of transfusion to prevent transfusion reactions. This generic type of device may include a BECS accessory, a device intended for use with BECS to augment the performance of the BECS or to expand or modify its indications for use (21 CFR 864.9165). ↩
-
See 21 CFR 860.3(c)(3). ↩
-
See 21 CFR 803.3(w). ↩
-
Example guidances that distinguish between “probable risks” and “purely hypothetical risks” include, but are not limited to: (1) Factors to Consider When Making Benefit-Risk Determinations for Medical Device Investigational Device Exemptions - Guidance for Investigational Device Exemption Sponsors, Sponsor-Investigators and Food and Drug Administration Staff available at: https://www.fda.gov/regulatory-information/search-fda-guidancedocuments/factors-consider-when-making-benefit-risk-determinations-medical-device-investigational-device; and (2) Factors to Consider When Making Benefit-Risk Determinations in Medical Device Premarket Approval and De Novo Classifications available at: https://www.fda.gov/regulatory-information/search-fda-guidancedocuments/factors-consider-when-making-benefit-risk-determinations-medical-device-premarket-approval-and-de. ↩
-
FDA may request additional architecture charts to address the cybersecurity risks associated with a device. Refer to the guidance document, Content of Premarket Submissions for Management of Cybersecurity in Medical Devices, for more information. ↩
-
If a device uses OTS software, FDA may request additional information in premarket submissions. Please refer to section VII.B of this guidance and the guidance document, Guidance for Off-The-Shelf Software Use in Medical Devices, for more information. ↩
-
More information on interoperable medical devices is available at: https://www.fda.gov/regulatoryinformation/search-fda-guidance-documents/design-considerations-and-pre-market-submission-recommendationsinteroperable-medical-devices. ↩
-
For purposes of the system and software architecture diagram, this guidance considers a module to be a discrete unit or architectural item within the system or software. A module could represent, for example, a finished hardware device within a system of hardware and software products, a hardware component within a finished hardware device, a finished software product within a system of software products, or a software function within a finished software product. A module is not specifically meant to describe code-level software functions, although such code-level software functions could be considered modules if appropriate. The sponsor should determine what constitutes a module in the context of its system and software. ↩
-
Available at http://www.imdrf.org/docs/imdrf/final/technical/imdrf-tech-140918-samd-framework-riskcategorization-141013.pdf. ↩
-
XXX denotes a unique number identifier for a specific hazard, software requirement specification, software design specification, unit test case, integration test case or system test case. ↩
-
The term “requirements” is used in this section as part of the term “Software Requirements Specification”, and does not refer to a regulatory requirement. ↩
-
Hardware requirements generally include, but are not limited to, requirements related to: microprocessors, memory devices, sensors, energy sources, safety features, and communications. ↩
-
Programming language requirements generally include, but are not limited to, requirements related to program size requirements or restrictions, and information on management of memory leaks. ↩
-
Software performance and functional requirements generally include, but are not limited to, requirements related to algorithms or control characteristics for therapy, diagnosis, monitoring, alarms, analysis, and interpretation with full text references or supporting clinical data, if necessary. Software performance and functional requirements may also include: device limitations due to software, internal software tests and checks, error and interrupt handling, fault detection, tolerance, and recovery characteristics, safety requirements, and timing and memory requirements. ↩
-
Interface requirements (e.g., external, user, internal) generally include, but are not limited to, both communication between system components and communication with the user such as: printers, monitors, keyboard, mouse, cloud servers, peripheral medical devices, mobile technology platforms. ↩
-
Available at https://www.fda.gov/regulatory-information/search-fda-guidance-documents/general-principlessoftware-validation. ↩
-
See 21 CFR 820.30(c) (“Design input. Each manufacturer shall establish and maintain procedures to ensure that the design requirements relating to a device are appropriate and address the intended use of the device, including the needs of the user and patient. The procedures shall include a mechanism for addressing incomplete, ambiguous, or conflicting requirements. The design input requirements shall be documented and shall be reviewed and approved by a designated individual(s). The approval, including the date and signature of the individual(s) approving the requirements, shall be documented.”). ↩
-
Available at https://www.fda.gov/regulatory-information/search-fda-guidance-documents/general-principlessoftware-validation. ↩
-
Available at https://www.fda.gov/regulatory-information/search-fda-guidance-documents/general-principlessoftware-validation. ↩
-
Available at https://www.fda.gov/regulatory-information/search-fda-guidance-documents/changes-existingmedical-software-policies-resulting-section-3060-21st-century-cures-act. ↩
-
Available at https://www.fda.gov/regulatory-information/search-fda-guidance-documents/general-wellnesspolicy-low-risk-devices. ↩
-
Available at https://www.fda.gov/regulatory-information/search-fda-guidance-documents/policy-device-softwarefunctions-and-mobile-medical-applications. ↩
-
Available at https://www.fda.gov/regulatory-information/search-fda-guidance-documents/medical-device-datasystems-medical-image-storage-devices-and-medical-image-communications-devices. ↩
-
Available at https://www.fda.gov/medical-devices/classify-your-medical-device/how-determine-if-your-productmedical-device. ↩
-
Please note “Basic Documentation” is only applicable to the Off-The-Shelf Software Use in Medical Devices (OTS) guidance and is NOT the same as “Basic Documentation Level.” “Basic Documentation” is described in the OTS guidance as the answers to questions pertaining to the description of OTS software; whereas “Basic Documentation Level” is described in this guidance as one of two levels of documentation recommendations used to help identify the minimum amount of information that, based on FDA’s experience, would generally be needed to support a premarket submission for a device that uses software. ↩
-
Available at https://www.fda.gov/regulatory-information/search-fda-guidance-documents/recognition-andwithdrawal-voluntary-consensus-standards. ↩
-
Available at https://www.fda.gov/regulatory-information/search-fda-guidance-documents/appropriate-usevoluntary-consensus-standards-premarket-submissions-medical-devices. ↩
