
OBSOLETE: Replaced by the 2023 Guidance 🔗
About this Transcript 🔗
This document is a transcript of an official FDA (or IMDRF) guidance document. We transcribe the official PDFs into HTML so that we can share links to particular sections of the guidance when communicating internally and with our clients. We do our best to be accurate and have a thorough review process, but occasionally mistakes slip through. If you notice a typo, please email a screenshot of it to Mihajlo at mgrcic@innolitics.com so we can fix it.
Preamble 🔗
Document issued on September 27, 2019.
For questions about this document, contact the Division of Digital Health by e-mail at digitalhealth@fda.hhs.gov.
I. Introduction 🔗
Off-the-shelf (OTS) Software is commonly being considered for incorporation into medical devices as the use of general-purpose computer hardware becomes more prevalent. The use of OTS Software in a medical device allows the manufacturer to concentrate on the application software needed to run device-specific functions. However, OTS Software intended for general-purpose computing may not be appropriate for a given specific use in a medical device. The medical device manufacturer using OTS Software generally gives up software life cycle control, but still bears the responsibility for the continued safe and effective performance of the medical device.
This guidance document was developed to address the many questions asked by medical device manufacturers regarding what they need to provide in a premarket submission to the FDA when they use OTS Software. The specific response to these questions depends on the medical device in question and the impact on patient, operator, or bystander safety if the OTS Software fails. Thus, the answer to the question, “What do I need to document?” may differ and is based on the risk analysis that is an integral part of designing a medical device. The detail of documentation to be provided to FDA and the level of life cycle control necessary for the medical device manufacturer increase as severity of the hazards to patients, operators, or bystanders from OTS Software failure increases.
This document lays out in broad terms how the medical device manufacturer can consider what is necessary to document for submission to the Agency. A basic set of need-to document items is recommended for all OTS Software, and a detailed discussion is provided on additional (special) needs and responsibilities of the manufacturer when the severity of the hazards from OTS Software failure become more significant.
For the current edition of the FDA-recognized standard(s) referenced in this document, see the FDA Recognized Consensus Standards Database.1 For more information regarding use of consensus standards in regulatory submissions, please refer to the FDA guidance titled Appropriate Use of Voluntary Consensus Standards in Premarket Submissions for Medical Devices.2”
FDA's guidance documents, including this guidance, do not establish legally enforceable responsibilities. Instead, guidances describe the Agency’s current thinking on a topic and should be viewed only as recommendations, unless specific regulatory or statutory requirements are cited. The use of the word should in Agency guidance means that something is suggested or recommended, but not required.
II. Scope 🔗
The purpose of this document is to describe the information that generally should be provided in a medical device application involving OTS Software. This information is in addition to the documentation described in the Guidance for the Content of Premarket Submissions for Software Contained in Medical Devices.3 Many of the principles outlined herein may also be helpful to device manufacturers in establishing design controls and validation plans for use of OTS Software in their devices. This guidance discusses key elements reviewers should look for in the submission, thereby providing a common baseline from which both manufacturers and reviewers can operate. This should improve predictability of Agency interaction with sponsors regarding applications involving OTS Software.
The guidance provided in this document reflects a safety-based approach to risk management and is designed to be consistent with international standards on risk management. Existing international standards indicate that the estimation of risk should be considered as the product of the severity of harm and the probability of occurrence of harm. Probabilities of occurrence are calculated based on clinical and engineering considerations. On the clinical side, manufacturers use patient populations, user skill sets, labeling, and risk benefit analysis to calculate risk and acceptable risk levels. On the software engineering side, probabilities of occurrence would normally be based on software failure rates. However, software failures are systematic in nature and therefore their probability of occurrence can not be determined using traditional statistical methods.
Because the risk estimates for hazards related to software cannot easily be estimated based on software failure rates, CDRH has concluded that engineering risk management for medical device software should focus on the severity of the harm that could result from the software failure. ‘Hazard Analysis’ is defined as the identification of hazards and their initiating causes. Based on the definition of ‘Risk Analysis,4’ hazard analysis is actually a subset of risk analysis; because risk analysis for software cannot be based on probability of occurrence, the actual function of risk analysis for software can then be reduced to a hazard analysis function. Technically speaking, the use of either term risk or hazard analysis is appropriate. However, CDRH has chosen to use the term hazard analysis to reinforce the concept that calculating risk based on software failure rates is generally not justified, and that it is more appropriate to manage software safety risk based on the severity of harm rather then the software failure rates.
III. Definitions 🔗
Following a safety-based approach to risk analysis, we define:
Hazard – A possible source of danger or a condition that could result in human injury.
Hazard Analysis – Identification of hazards and their initiating causes.
Hazard Mitigation – Reduction in the severity of the hazard, the likelihood of the occurrence, or both.
Major Level of Concern – The Level of Concern is major if operation of the software associated with device function directly affects the patient, operator, and/or bystander so that failures or latent flaws could result in death or serious injury to the patient, operator, and/or bystander, or if it indirectly affects the patient, operator, and/or bystander (e.g., through the action of care provider) such that incorrect or delayed information could result in death or serious injury to the patient, operator, and/or bystander.
Minor Level of Concern – The Level of Concern is minor if failures or latent design flaws would not be expected to result in any injury to the patient, operator, and/or bystander.
Moderate Level of Concern – The Level of Concern is moderate if the operation of the software associated with device function directly affects the patient, operator, and/or bystander so that failures or latent design flaws could result in non-serious injury to the patient, operator, and/or bystander, or if it indirectly affects the patient, operator, and/or bystander (e.g., through the action of the care provider) where incorrect or delayed information could result in non-serious injury of the patient, operator, and/or bystander.
Off-the-Shelf Software (OTS Software) – A generally available software component, used by a medical device manufacturer for which the manufacturer cannot claim complete software life cycle control.
Risk Analysis – Investigation of available information to identify hazards and to estimate risks.5
Risk Control – The process through which decisions are reached and implemented for reducing risks to, or maintaining risks within, specified limits.6
Safety – In the regulation of medical devices, safety means that the probable benefits to health for its intended use when accompanied by adequate directions and warnings against unsafe use, outweigh any probable risks. In this guidance we will use the words “safety and effectiveness” to remind ourselves that safety is only meaningful in the context of the benefit-risk considerations and the labeling.
Serious Injury – As adopted from the Medical Device Reporting (MDR) regulation in 21 CFR 803.3(w), means an injury or illness that:
- Is life threatening,
- Results in permanent impairment of a body function or permanent damage to a body structure, or
- Necessitates medical or surgical intervention to preclude permanent impairment of a body function or permanent damage to a body structure.
Permanent – For the purpose of this subpart, permanent means irreversible impairment or damage to a body structure or function excluding trivial impairment or damage.
Other software terminology used in this document is defined in FDA's Glossary of Computer System Software Development Terminology.7
IV. OTS Software Decision Schematic 🔗
The content of the application supporting use of OTS Software in a medical device depends on the results of the hazard analysis. Figure 1 provides a schematic of the decision process and a table of contents for Section V of this guidance document.
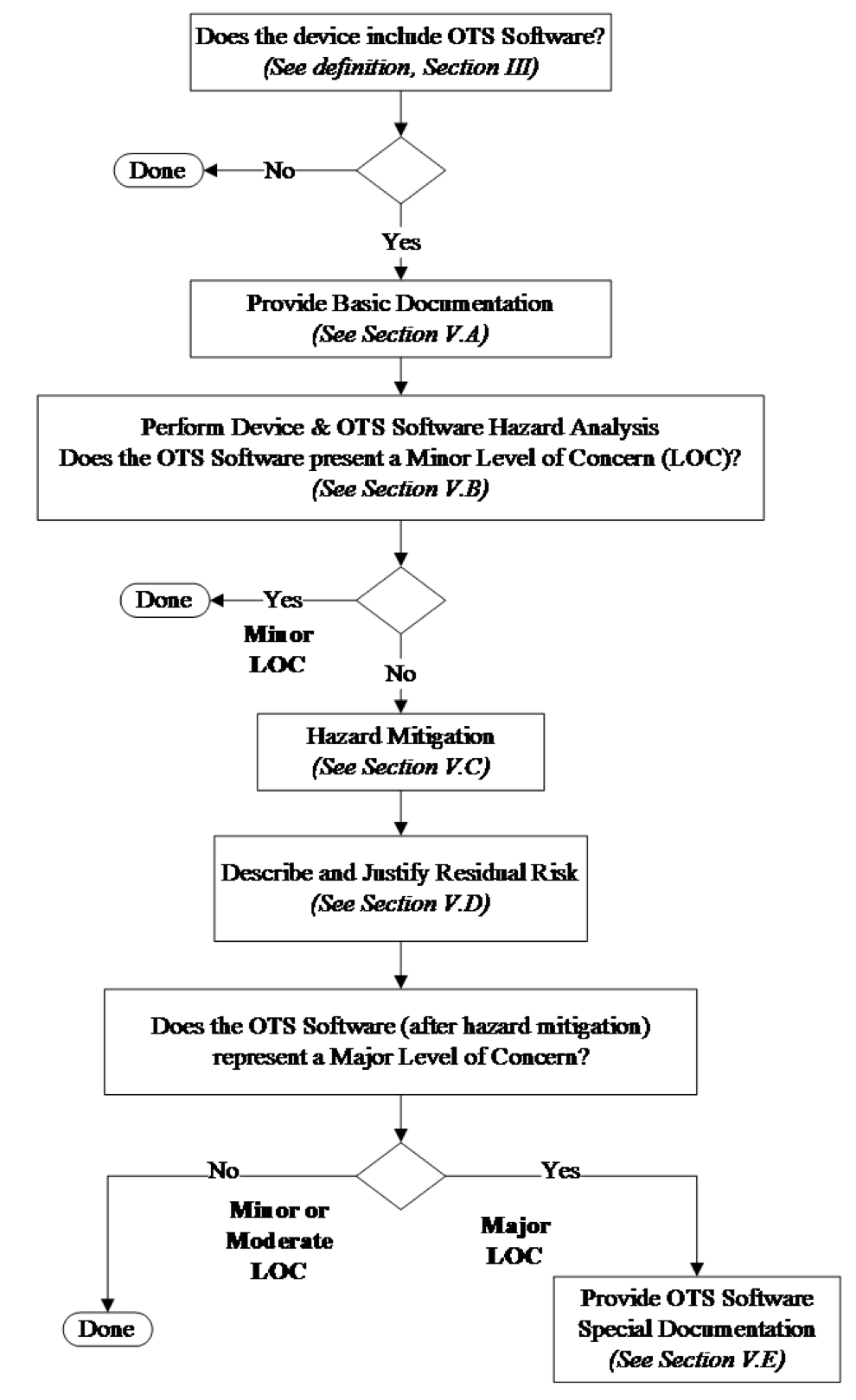
Table 1 summarizes the recommended contents for an OTS Software submission based on Figure 1.
Table 1. Documentation Summary from Figure 1.
| Minor Level of Concern before mitigations | Minor Level of Concern after mitigations | Moderate Level of Concern | Major Level of Concern after mitigations |
|---|---|---|---|
| Hazard Analysis | Hazard Analysis | Hazard Analysis | Hazard Analysis |
| Basic Documentation | Basic Documentation | Basic Documentation | Basic Documentation |
| Hazard Mitigations | Hazard Mitigations | Hazard Mitigations | |
| Describe and Justify Residual Risk | Describe and Justify Residual Risk | ||
| Special Documentation |
V. OTS Software Use 🔗
A. Basic Documentation for OTS Software 🔗
The OTS Software Basic Documentation is intended to answer the following questions:
-
What is it? For each component of OTS Software used, the following should be specified:
- Title and Manufacturer of the OTS Software.
- Version Level, Release Date, Patch Number, and Upgrade Designation, as appropriate.
- Any OTS Software documentation that will be provided to the end user.
- Why is this OTS Software appropriate for this medical device?
- What are the expected design limitations of the OTS Software?
Note: The medical device manufacturer should only use the OTS Software as specified in an appropriate document, i.e., design record. If the version of the OTS Software changes, the appropriate document should be updated to reflect the change.
-
What are the Computer System Specifications for the OTS Software? For what configuration will the OTS Software be validated? The following should be specified:
- Hardware specifications: processor (manufacturer, speed, and features), RAM (memory size), hard disk size, other storage, communications, display, etc.
- Software specifications: operating system, drivers, utilities, etc. The software requirements specification (SRS) listing for each item should contain the name (e.g., Windows 10, Excel, Sun OS, etc.), specific version levels (e.g., 4.1, 5.0, etc.) and a complete list of any patches that have been provided by the OTS Software manufacturer.
-
How will you assure appropriate actions are taken by the End User?
- What aspects of the OTS Software and system can (and/or must) be installed/configured?
- What steps are permitted (or must be taken) to install and/or configure the product?
- How often will the configuration need to be changed?
- What education and training are suggested or required for the user of the OTS Software?
- What measures have been designed into the medical device to prevent the operation of any non-specified OTS Software, e.g., word processors, games? Operation of non-specified OTS Software may be prevented by system design, preventive measures, or labeling. Introduction may be prevented by disabling input (USB, CD, modems).
-
What does the OTS Software do? What function does the OTS Software provide in this device? This is equivalent to the software requirements in the Guidance for the Content of Premarket Submissions for Software Contained in Medical Devices8 for this OTS Software. The following should be specified:
- What is the OTS Software intended to do? The sponsor’s design documentation should specify exactly which OTS components will be included in the design of the medical device and to what extent OTS Software is involved in error control and messaging in device error control.
- What are the links with other software including software outside the medical device (not reviewed as part of this or another application)? The links to outside software should be completely defined for each medical device/module. The design documentation should include a complete description of the linkage between the medical device software and any outside software (e.g., networks).
-
How do you know it works? Based on the Level of Concern:
- Describe testing, verification, and validation of the OTS Software and ensure it is appropriate for the device hazards associated with the OTS Software. (See Note 1.)
- Provide the results of the testing. (See Note 2.)
- Is there a current list of OTS Software problems (bugs) and access to updates?
Note 1: FDA recommends that software test, verification, and validation plans identify the exact OTS Software (title and version) that is to be used. When the software is tested it should be integrated and tested using the specific OTS Software that will be delivered to the user.
Note 2: If the manufacturer allows the use of the medical device with different versions of OTS Software, then the manufacturer should validate the medical device for each OTS Software version.
-
How will you keep track of (control) the OTS Software? An appropriate plan should answer the following questions:
- What measures have been designed into the medical device to prevent the introduction of incorrect versions? On startup, ideally, the medical device should check to verify that all software is the correct title, version level, and configuration. If the correct software is not loaded, the medical device should warn the operator and shut down to a safe state.
- How will you maintain the OTS Software configuration?
- Where and how will you store the OTS Software?
- How will you ensure proper installation of the OTS Software?
- How will you ensure proper maintenance and life cycle support for the OTS Software?
B. OTS Software Hazard Analysis 🔗
A comprehensive risk management approach includes hazard analysis and mitigation that continues iteratively throughout the life of the product. The manufacturer is expected to perform an OTS Software hazard analysis as a part of a medical device (system) hazard analysis.
OTS Software failure, malfunction, or misuse may present a hazard to the patient, operators, or bystanders. Figure 2 summarizes the typical hazard management and mitigation process that would include a hazard analysis of the OTS Software component.
The submission should include the following information to document the OTS Software hazard analysis:
- A list of all potential hazards identified.
- The estimated severity of each identified hazard.
- A list of all potential causes of each identified hazard.
Note: A tabular format of the OTS Software hazard analysis or a tabular summary will facilitate review. The hazard analysis for OTS Software may be included in the overall device hazard analysis provided adequate documentation is provided.
If the device with the OTS Software represents a Minor Level of Concern, then the Level of Concern for the OTS Software can be no greater. The hazard analysis for the OTS Software in such a device may simply document the Minor Level of Concern of the device.
Where failure, malfunction, or misuse of the OTS Software poses no possibility of injury to the patient, operators, or bystanders, then the OTS Software is said to present a Minor Level of Concern, and the fulfillment of the Basic Documentation (see Section V.A) will be considered sufficient.
C. OTS Software Hazard Mitigation 🔗
Hazard mitigation activities may seek to reduce the severity of the hazard, the likelihood of the occurrence, or both. Hazard mitigation interventions may be considered in three categories with the following order of precedence:
- Design (or redesign)
- Protective measures (passive measures)
- Warning the user (labeling)
Figure 2. Typical Hazard Analysis and Mitigation
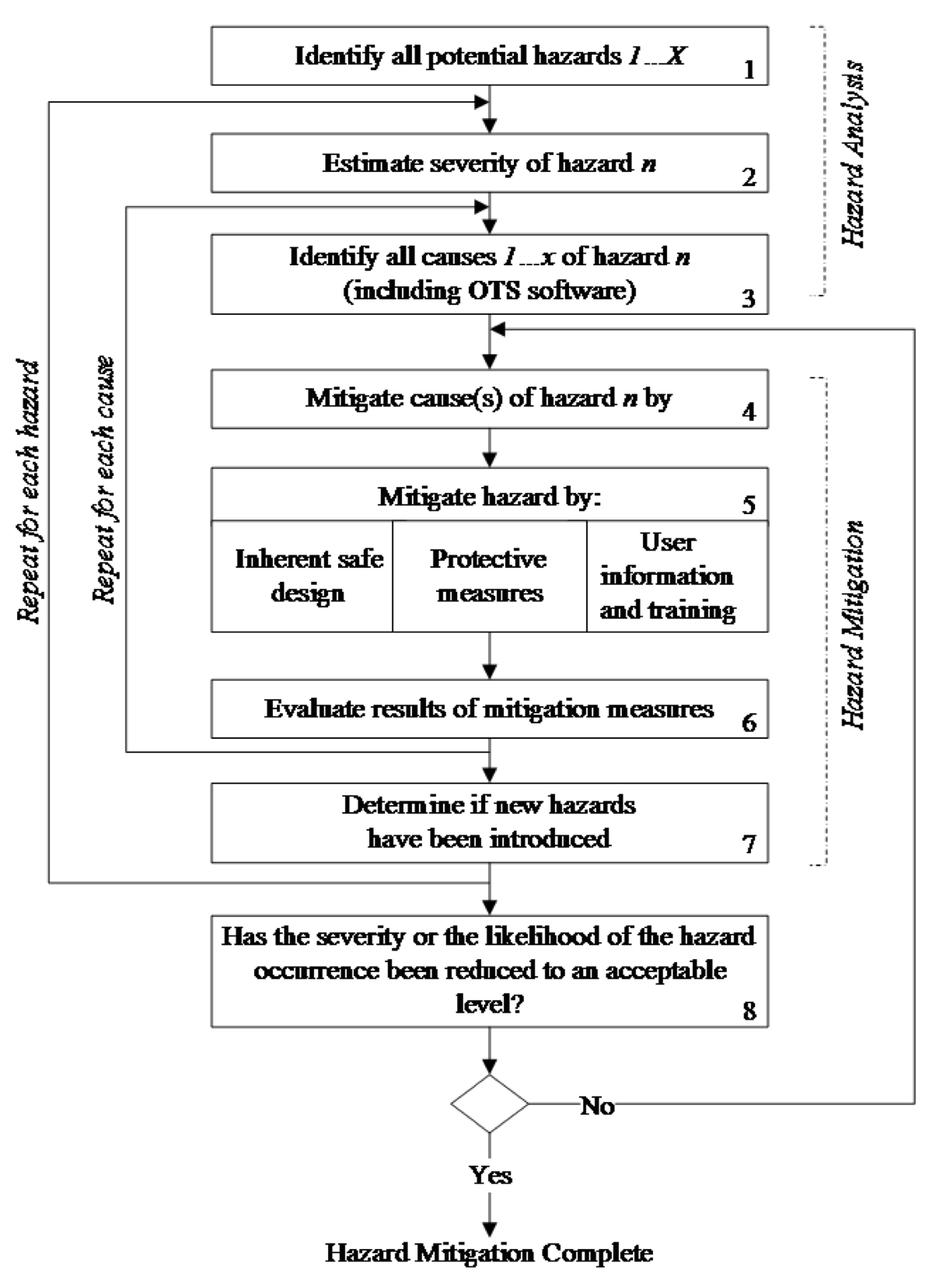
These approaches may involve hardware and/or software. These three mitigation approaches are by no means mutually exclusive and may be used concurrently. The most desirable approach is to design in effective controls, i.e., eliminate the need for a hazardous operation or component. Protective measures are considered passive (from the user’s standpoint) since they do not require any action on the part of the user. The least effective approaches depend on some action (or lack of action) on the part of the medical device user.
The submission should include the following information to document the OTS Software hazard mitigation:
- A list of all identified medical device hazards associated with the OTS Software;
- The steps taken to mitigate each hazard; and
- The residual risk.
Note: A tabular format of the risk management or a tabular summary will facilitate review. These results will typically be included as a part of the overall medical device hazard analysis and mitigation plan.
One example of a comprehensive approach to injury prevention in public health was developed around ten “countermeasures.9” Table 2 (see next page) illustrates a generic approach to the hazard mitigation, in this case, to preventing injury-related energy release to patients, operators, or bystanders.
With implementation of each hazard mitigation, the residual risk is assessed as well as assessment of any new hazards that may be introduced.
Acceptable levels of residual risk, based on the severity or the likelihood of the residual risk occurring, will depend on the intended use of the medical device and the function performed by the software. In the case of diagnostic tests, injury includes results that can lead to unnecessary invasive diagnostic testing (e.g., biopsy) or withholding or delaying important diagnostic or therapeutic procedures.
The sponsor will need to describe and justify the residual risk (Section V.D) for Moderate or Major Levels of Concern. Where failure, malfunction, or misuse of the OTS Software is likely to result in death or serious injury to the patient, operators, or bystanders, then the OTS Software is said to present a Major Level of Concern. If the residual risk from the OTS Software presents a Major Level of Concern, the sponsor will need to fulfill Special Documentation (Section V.E).
Table 2. Injury Reduction Countermeasures
| Countermeasure |
|---|
| 1. Prevent accumulation of the energy. |
| 2. Reduce the amount of the energy delivered. |
| 3. Prevent inappropriate release of the energy. |
| 4. Modify the release of the energy. |
| 5. Separate the patient from the energy in time and space. |
| 6. Provide physical barriers between the energy and the patient. |
| 7. Change the surfaces or basic structures at the interface. |
| 8. Reduce likelihood of misapplication or Increase resistance of the patient. |
| 9. Provide rapid emergency response to injury. |
| 10. Improve medical care and rehabilitation after the injury. |
D. Describe and Justify Residual Risk 🔗
The sponsor should provide a detailed (complete) discussion of the risk that remains.
The risk related to the use of OTS Software should be considered in relation to the risk of the alternatives, e.g., custom developed software. Any experience (data) with the use of the OTS Software in this or a related application should be presented by the sponsor and will be considered by the reviewers. Whether the residual risk is acceptable depends on the specific medical device application.
E. Special Documentation for OTS Software 🔗
To fulfill Special Documentation for OTS Software of a Major Level of Concern, the medical device manufacturer is expected to:
- Provide assurance to FDA that the product development methodologies used by the OTS Software developer are appropriate and sufficient for the intended use of the OTS Software within the specific medical device. FDA recommends this include an audit of the OTS Software developer’s design and development methodologies used in the construction of the OTS Software. This audit should thoroughly assess the development and qualification documentation generated for the OTS Software (see Note below). Note: If such an audit is not possible and after hazard mitigation, the OTS Software still represents a Major Level of Concern, the use of such OTS Software may not be appropriate for the intended medical device application.
- Demonstrate that the procedures and results of the verification and validation activities performed for the OTS Software are appropriate and sufficient for the safety and effectiveness requirements of the medical device. Verification and validation activities include not only those performed by the OTS Software developer, but also include those performed by the medical device manufacturer when qualifying the OTS Software for its use in the specific medical device.
- Demonstrate the existence of appropriate mechanisms for assuring the continued maintenance and support of the OTS Software should the original OTS Software developer terminate their support.
VI. OTS Software Used in Marketing Applications 🔗
A. Examples 🔗
Examples of medical devices using OTS Software are described in this section. These examples illustrate the reasoning that leads to defining the Level of Concern for a medical device and thus the kinds of development processes that should be used and the information to be provided in a regulatory submission.
(1) Corneal Topographer 🔗
Minor Level of Concern medical device (see Section V.A).
Intended Use: A corneal topographer provides images of the abnormalities in the curvature of the cornea, the simplest being astigmatism.
Description: A corneal topographer consists of a hollow cone that the patient looks into from the base looking towards the interior of the point (like looking into the big end of a megaphone with one eye). The inside of the cone is white with black concentric circles. The concentric circles reflect off the eye and are imaged by a camera with a computer controlled lens situated at the point of the cone looking at the patient’s eye. The shapes of the reflections of the concentric circles are used to develop a topographic map of the cornea curvature that is printed out.
OTS Software: An OTS operating system such as Windows is commonly used to interface the user, the microcomputer hardware platform, the corneal topographer, data storage, and output devices.
OTS Software Level of Concern: A corneal topographer represents no threat of direct harm to the patient. The risk of indirect harm from a misdiagnosis relating to medical device malfunction is small since the worst case is an incorrect image that is considered correct. The OTS Software in this medical device thus represents a Minor Level of Concern (see Section V.B) and should satisfy Basic Documentation (see Section V.A).
(2) Perineometer 🔗
Minor Level of Concern medical device (see Section V.A).
Intended Use: Perineometers are used to provide feedback to a patient performing muscle strengthening exercises (Kegel exercises) for the treatment of certain types of urinary incontinence.
Description: There are two types of perineometers: those that measure pressure, and those that measure electrical activity (EMG) from muscles. Each device consists of a probe that is placed into either the vagina or the rectum, and a monitoring unit. The pressure devices use an air-filled probe connected to the monitoring unit by a piece of plastic tubing. When the patient performs the exercise, the probe is compressed, and the monitoring unit reports the change in pressure. The electrical devices use an electrode to measure the electrical activity of the target muscles during the exercises, and this information is reported by the monitoring unit.
OTS Software: An OTS operating system, such as DOS or Windows, may be used to record and display the data collected by the monitoring unit.
OTS Software Level of Concern: Perineometers represent no threat of direct injury to the patient, since no energy is applied by the medical device to the patient. The risk of indirect injury due to inaccurate feedback during the exercise session is expected to be small, as these medical devices are only used as an adjunct to exercise therapy, and they are used under clinical supervision. The OTS Software in this medical device thus represents a Minor Level of Concern (see Section V.B) and should satisfy the Basic Documentation (see Section V.A).
(3) Implantable Medical Device Programmers 🔗
Describe and Justify Residual Risk (see Section V.E).
Intended Use: An implantable medical device programmer provides interface and two-way communication with an implantable cardioverter-defibrillator (ICD) or cardiac pacemaker.
Description: An implantable medical device programmer consists of an electromagnetic programming head that is placed over the implanted device and provides through-the-skin communication with the implanted device, the personal computer (PC) interface, and the PC hardware and software. The programmer permits the physician-user to:
- Query the implant for performance history (device and patient), and, in some systems, for print-out of the recorded electrograms;
- Set the adjustable (programmable) characteristics of the implant;
- Provide the induced shock for system initialization and diagnostic purposes; and · Verify implant operating characteristics and status (including battery) via signals from the implant.
OTS Software: An OTS operating system such as DOS or Windows is used to provide a user interface (sometimes graphical), interface to the PC (hardware platform), and interface with data storage, and output devices.
OTS Software Level of Concern: The on-board software for the implant satisfies the definition of Major Level of Concern software (life supporting/life sustaining) and would need to satisfy the Special Documentation (see Section V.E). Whether the device programmer can be considered of lesser Level of Concern depends primarily on the protection designed into the implant or the programmer. Steps taken to mitigate the risk might include:
- Design of the implant to minimize the possibility of misprogramming to inappropriate operational states;
- Design of the programmer interface to minimize the chance of miscommunication, including hardening of the hardware against electromagnetic interference (EMI);
- Limiting the part of the OTS Software that is utilized in the programming application;
- Protecting the PC from use for other applications, including consideration of the following:
- Software design features to protect against adding unwanted software, modification, or system use; and
- Hardware design features to protect against unwanted system use.
Other points that might be offered to support use of OTS Software in the programmer might include:
- Documented experience (data) with use of the OTS Software in this application.
- What was the system in place to detect and report problems?
- What is the rate of problems reported compared to other (perhaps non-OTS Software) systems?
- Documented experience with the OTS Software in other relevant applications.
- What are the reported problems (bug list) and how many are relevant to this application?
- Has there been difficulty in developing work-arounds for the problems relevant to this application?
The review team must decide whether the overall programmer system as implemented satisfies the necessary system safety and effectiveness (see Section V.E).
B. 510(k) Issues with OTS Software 🔗
The conditions under which a new or changed medical device including OTS Software will require a new 510(k) are the same as for a device not involving OTS Software. These conditions are given in the CDRH’s guidances, “Deciding When to Submit a 510(k) for a Change to an Existing Device,10 “ and “Deciding When to Submit a 510(k) for a Software Change to an Existing Device.11 “
(1) OTS Software Changes Requiring a 510(k) 🔗
For medical devices where the OTS Software represents a Minor Level of Concern, OTS Software changes would not typically require a new 510(k). However, the manufacturer is responsible for validating the change.
For other medical devices, the decision as to whether a new 510(k) is required depends on the intended use of the device; the function of the OTS Software; and to what extent the risks due to OTS Software have been mitigated (see guidances on when to submit a 510(k) for a modification12𝄒13).
C. Investigational Device Exemption Issues with OTS Software 🔗
The requirements for an investigational device exemption (IDE) are the same whether or not the medical device contains OTS Software. The OTS Software may be a component of a medical device or the OTS Software may be the entire medical device, e.g., diagnostic software. The conditions that would require submission of an IDE are specified in section 21 CFR 812 and generally include changes that would affect the patient population for which the medical device is intended; conditions of use of the device (including those recommended or suggested in the labeling or advertising; the probable benefit from the use of the device weighed against any probable injury or illness from such use); or the reliability of the medical device.
Some specific issues related to OTS Software might include initial (beta) testing of an OTS Software medical device in clinical studies. Such a study must comply with applicable IDE requirements.14 For non-significant risk medical devices, that includes approval by an institutional review board (IRB) and patient informed consent. For significant risk studies, the initial user testing (beta testing) protocol would be included in an IDE submission. For example, beta testing of radiation treatment planning software, including any OTS Software modules, would be conducted under a full IDE with FDA approval as a prerequisite. See the guidance on “Significant Risk and Nonsignificant Risk Medical Device Studies15” for more information.
D. Exemption of Certain Diagnostic Devices 🔗
If the product incorporating the OTS Software is a diagnostic medical device, it may be exempted from IDE requirements, if it meets the criteria in section 21 CFR 812.2(c)(3). For example, clinical (beta) testing of a noninvasive diagnostic device that does not require significant risk invasive sampling procedure and that does not introduce energy into the body, is exempted from IRB approval, patient informed consent, and other IDE requirements, if a medically established diagnostic product or procedure is used to confirm the diagnosis.
E. Premarket Approval Issues with OTS Software 🔗
The criteria and requirements for premarket approval (PMA) applications are in section 21 CFR 814. When a manufacturer submits a PMA submission for a medical device, there must be valid scientific evidence (including clinical evidence, if needed) to support a reasonable assurance of safety and effectiveness of the device.16
The OTS Software used in a medical device is evaluated in the context of the overall medical device. The extent to which the medical device manufacturer ensures that the OTS Software was developed using appropriate life cycle control depends upon the overall risk of the medical device, the role of the OTS Software, and the Level of Concern associated with possible failures of the OTS Software component.
For example, a commercially available neural network, used by a medical device manufacturer for pattern recognition, would require extensive validation if used in a Pap smear screening device, in computer-assisted radiology, or for computer-assisted analysis of ECG waveforms. The same neural network, used for less critical computer-assisted analysis of EEG waveforms, might require less rigorous software documentation. Likewise, a commercially available personal computer operating system with graphical user interface, would require extensive documentation and evidence of validation when intended for use in a cardiac pacemaker programmer. Less documentation and verification of the OTS operating system would be required for programming an artificial ear.
F. Artificial Intelligence 🔗
OTS knowledge-based software (for example, artificial intelligence, expert systems, and neural net software) are being developed for a number of medical applications. A typical system accepts clinical findings (sometimes including imaging data) and generates probabilities of disease states and/or recommendations for subsequent data gathering or treatment. The clinician may order a surgical biopsy or other invasive tests or initiate therapy based on the system output. Such systems should be tested and reviewed in a manner consistent with both their safety and effectiveness of their direct effects (recommendations) and indirect effects (missed appropriate diagnostic testing and treatment).
G. Product Labeling 🔗
FDA recommends that the user’s manual specify the version(s) of the OTS Software that can be used with the medical device. Such specification would not be needed for embedded software (i.e., the user does not select the OTS Software and cannot change the software provided by the medical device manufacturer).
The user’s manual should contain appropriate warnings to the user indicating that the use of any software other than those specified will violate the safety, effectiveness, and design controls of this medical device and that such use may result in an increased risk to users and patients. Further description of what comprises a warning and how to write it are included in the Device Labeling Guidance17 and the guidance on Labeling – Regulatory Requirements for Medical Devices.18
When OTS medical device software is delivered on a magnetic / user installable medium, the package should include labeling that indicates the minimum hardware platforms on which the software is validated to run (processor, memory, disk, interface, etc.). The appropriate testing for the user to assure proper installation should also be described in the labeling.
If the hardware on which the OTS Software runs is a stand-alone computer and the user is not “locked out” by hardware or software system features, then the user should be warned against installing any other software (utilities or applications programs) on the computer.
VII. Appendix 🔗
The purpose of these Appendices is to provide background and comment on various OTS Software. Based on the Level of Concern, device manufacturers should either use or not use Commercial Off-the-shelf Software (COTS).
A. Operating Systems 🔗
The operating system software is the primary software program that manages the basic functions of the computer and its associated hardware, including peripherals. The operating system provides a basic user interface, is responsible for managing applications programs and tasks, controlling memory allocation and data storage devices, and providing input/output for the computer, as well as any additional peripheral devices that are present.
“Open” hardware (mass market) architecture computers vary widely in architectural and organizational characteristics such as timing, addressing, and processing. Operating systems and application software executing on these platforms should be “robust” enough to perform appropriately in this environment.
OTS driver software packages provide interface functions between the CPU, operating system, and the input/output peripheral. However, the performance and functionality of the OTS driver software may be affected by the overall system configuration and the OTS hardware. In general, OTS driver software packages can be classified into the following input/output interface types: serial, parallel, video signal, telemetry, LAN, and internal bus.
In most cases, a particular software driver derives from a particular interface protocol and contains the data signals, control signals, and timing signals for proper operation.
Since tests for most input/output interface/bus configurations require the particular bus analysis or logic analysis, scope, and knowledge of the particular interface protocol, the validation process for the OTS driver software package should be part of the system interface validation process for higher levels of concern. This includes the verification of the data values in both directions for the data signals; various mode settings for the control signals in both directions (if applicable); and the input/output interrupt and timing functions of the driver with the CPU and operating system.
B. Utilities and Drivers 🔗
The purpose of this Appendix is to provide general recommendations and background for the use of OTS utility and driver software packages in the medical device validation process.
Utility software is generally designed to work with a specific operating system. Unlike applications software, utility software is intended to supplant or enhance functions typically performed by the operating system. Examples of utility programs are memory managers, file managers, and virus checkers. Networking software can also be considered as utility software in that it allows multiple computers to access the same resources. Operating systems can also be designed to support or enable network operations without any additional utility software.
OTS operating systems are commonly considered for incorporation into medical devices as the use of general-purpose computer hardware becomes more prevalent. The use of OTS operating system software allows device manufacturers to concentrate on the application software needed to run device-specific functions. However, an OTS operating system software is intended for general-purpose computing and may not be appropriate for a given specific use in a medical device. Developers of OTS operating systems typically design their systems for general-purpose business or consumer computing environments and tasks where software failures and errors are more accepted. This acceptability of errors in the general-purpose computing environment may make the OTS operating system software inappropriate for less error-tolerant environments or applications.
The incorporation of OTS operating system software may also introduce unnecessary functions and complexity into a medical device. General-purpose functional requirements typically result in the OTS operating system software being large and unwieldy in the attempt to incorporate more functionality into the operating system. This excess functionality is typically never used for specific medical device applications and increases the likelihood that errors may be introduced into the operating system. The basic functions of an OTS operating systems used for medical device applications are typically the graphical user interface environment and the hardware interface functions. There are a number of operating systems used for timing- or resource-critical applications that provide the basic functionality needed to support user and hardware interfaces, but do not have many of the disadvantages of general-purpose business or consumer operating systems.
OTS utility software packages can perform the following functions: math functions (fast Fourier transform, sin, cos); display functions (graphic); management functions (copy, delete, store various computer data/files); and the data manipulation function (transfer from one Boolean type or both). The validation for these types of the software should be appropriate to the Level of Concern.
C. Local Area Networks (LANs) 🔗
The purpose of this Appendix is to provide general recommendations and background for the network aspects of OTS Software use. Medical devices, particularly multi-parameter patient monitors and imaging systems, are increasingly networked for clinical work groups, centralized monitoring, and storage of patient medical data and records. LANs and other networks support more and more communication and sharing of images, measurement data, audio, video, graphics, text, etc. This heterogeneous media environment comes at a cost of more processing power, higher bandwidth or network speed, sophisticated object-relational databases, and security and access considerations.
The evaluation of networked medical devices begins with a definition of the technical requirements of the network application and the understanding of those requirements.
(1) Requirements Analysis 🔗
- Speed – The response time required for safe and effective operation determines the LAN data rate (bandwidth) for the medical device system. The CPU processing power and clock speed required at device monitors, workstations, and client machines should be appropriate so that bottlenecks do not occur.
- LAN Architecture – The size of the LAN (the number of user nodes) and the topology of the LAN should be specified.
- Discuss to what extent the LAN needs to be fault tolerant, e.g., when a workstation fails?
- Discuss to what extent the LAN needs to be scaleable, i.e., can new user nodes be added without degrading system performance?
- Discuss to what extent the main device software needs to be computationally selfsufficient or distributed.
- Network Operating System (NOS) – Whether OTS or proprietary, this selection should consider the trade-off between robustness and flexibility.
- Data Integrity – One of the most important issues for any medical device operating in a network is data integrity. The manufacturer should ensure that the network system software and hardware incorporate error checking, handling, and correction measures commensurate with the Level of Concern of the device.
- Transmission of data packets and files should include error detection and correction. Error detection methods include parity, checksum, and cyclic redundancy check (CRC).
- Transaction rollback after non-committed changes or network failure, supports data integrity in medical device LANs.
- Critical data and files may be stored in duplicate at separate locations.
- Network Management and Security – User authorization and authentication should precede accesses to sensitive patient information.
The above five items are not independent. Decisions made in one item area may affect the performance of the LAN in another area.
(2) Implementation 🔗
The speed required by the medical device system dictates the hardware selection, the network interface cards and transmissions protocols. For example, if the conventional Ethernet protocol (maximum transmission speed of 10 Mbps) is too slow for the intended application, then a different transmission protocol will be needed.
Simplicity of the LAN architecture versus fault tolerance is a trade-off that may arise in the implementation of the networked medical device systems. The LAN could be implemented as a linear bus network (perhaps the simplest scheme), but if any connecting link on the bus fails, the whole network can fail. A star topology with redundant centralized hub is an example of a more complex but more robust network structure.
Segmentation of high bandwidth applications may be employed to improve LAN performance. Limiting the data traffic to data intensive clusters reduces traffic throughout the overall LAN.
D. Device Master Files 🔗
Much of the information regarding development and validation of OTS Software may not be readily available to the medical device manufacturer who wishes to use the OTS Software as a device component. Commercial OTS Software vendors who wish to make their OTS Software available for use in medical devices, but do not want to share the confidential and/or proprietary details of their software development and validation with customers (medical device manufacturers), may direct the information in a device master file to the FDA. Additional information on Device Master Files can be found on the FDA website: https://www.fda.gov/medical-devices/premarket-approval-pma/master-files.
E. Maintenance and Obsolescence 🔗
This Appendix addresses relevant maintainability issues with regard to OTS Software in medical devices.
Maintenance activities are generally considered to begin subsequent to the establishment and distribution of a medical device product baseline. The distinction between maintenance and product development is an important one. Product development design activities generally lead to a system structure of highly integrated components and logic. Maintenance activities introduce changes into this structure that may lead to a loss in the integrity of the structure. Structure integrity may be affected through changes due to new design requirements, corrections, or environmental adaptations. These types of changes may impact the integrity of the structure organization, architecture, logic, integration, or any combination of these characteristics. Maintenance of products with OTS Software components may be particularly problematic for reasons discussed in the main body of this document, i.e., the sponsor does not have control of the OTS Software component life cycle process.
In particular, this section identifies general safety and effectiveness, design, verification / validation, change, installation, and decommissioning concerns. These concerns may be applied to all regulated medical device software and stand-alone medical software devices. The appropriate evaluation will depend on the Level of Concern.
Assumptions for this Section include:
- Manufacturer Good Software Development Practices (GSDP) and Good Corrective Action Practices (GCAP) are in place.
- A product baseline exists.
- A new product baseline based on a prior product baseline is under CDRH review.
Each concern below corresponds to a product development life cycle phase. The concerns identify fundamental maintenance concerns relevant to medical devices that include software. Guidance in the main body of this document provides the procedural foundation for concerns in this Section.
(1) Safety 🔗
Introduction of new or modified OTS components to a product baseline may impact the safety of the product. Therefore a safety impact assessment of the medical device should be performed and associated hazards documented in a Failure Modes and Effects Analysis (FMEA) table. Each hazard’s consequence should be provided and expressed qualitatively; e.g. major, moderate, or minor. Traceability between these identified hazards, their design requirements, and test reports should be provided.
Analysis should include the review of release bulletins (known error reports), user manuals, specifications, patches, and literature and internet searches for other user’s experience with this OTS Software. The submission should answer the following questions:
- Has a FMEA with traceability to requirements and test reports been provided?
- Are safety functions isolated from new OTS component(s)?
- Does the new OTS component affect system safety integrity?
- What new human factors conditions are introduced with new OTS components?
(2) Design 🔗
Introduction of new or modified OTS Software components to a product baseline may impact the original design of the product. This impact may result from necessary changes to the product structure organization, architecture, logic, integration, or a combination of these characteristics. Problems attributable to structural changes include:
- New system resource requirements, such as shared and/or fixed memory;
- New timing considerations;
- New memory organization (e.g., 16 bit to 32 bit to 64 bit words), partitioning;
- New human factor issues;
- New data integrity issues; and
- New software required to create the final code (build tools).
Consequently the submission should answer the following questions:
- How will the new OTS Software component(s) change the performance characteristics?
- How will the new OTS Software component(s) change the operational environment?
- Is data integrity preserved?
(3) Verification and Validation 🔗
As in the establishment of a product baseline, verification and validation (V&V) activities should occur when maintenance changes are made to a product baseline. Analysis of these changes directs necessary V&V activities. New OTS Software components in a product baseline introduce unknown logic paths and complexities into the product. “Black-box” testing of OTS Software components may allow some validation claims to be made. However, the unknown logic paths and complexities of OTS Software components make it important to know that design structure or logic elsewhere in the system is not impacted. This means a full system regression test should be performed. Results of these validation activities should be documented.
The submission should answer the following questions:
- Do test reports provide objective evidence that identified OTS Software component hazards have been addressed?
- Do test reports provide objective evidence that all identified system hazards have been addressed?
- Has a system regression test been performed?
(4) Installation 🔗
Changes in a product baseline structure resulting from the integration of new OTS Software components may impact installation requirements. This impact can range from minor documentation changes to field upgrades. The reviewer should ascertain the impact of OTS Software component changes on fielded products.
The submission should answer the following question: What is the impact of new OTS Software components on fielded medical device products?
For example: Do new OTS Software components correctly operate within the specifications of medical devices currently fielded?
(5) Obsolescence 🔗
Rapid technology changes, economics, and market demand are shrinking product life spans. A direct consequence of these phenomena is that an OTS Software component today may not exist two years from now. Short life spans are a particular characteristic of software because it is relatively easy to change. Obsolescence of OTS Software components can have significant impact on regulated products because the device manufacturer may lose the ability to properly support fielded products. The sponsor needs to support fielded medical device products with OTS Software components.
The submission should answer the following questions:
- Will the old OTS Software component still be available for fielded medical devices?
- Is there a retirement plan for OTS Software components to be replaced/eliminated?
- Do new OTS Software component(s) replace fielded components?
(6) Change Control 🔗
The submission must identify the product to be considered. Therefore, the product configuration provided should specify:
- Hardware platform (e.g. microprocessor, minimum memory required, addressable word size);
- Software platform (e.g. operating system, communications, databases, necessary utilities, etc.);
- OTS component(s) other than (B) above (see basic requirements in the main body of this document); and
- Internally developed application(s).
Footnotes 🔗
-
Available at https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfStandards/search.cfm. ↩
-
Available at https://www.fda.gov/regulatory-information/search-fda-guidance-documents/appropriate-usevoluntary-consensus-standards-premarket-submissions-medical-devices. ↩
-
Available at https://www.fda.gov/regulatory-information/search-fda-guidance-documents/guidance-contentpremarket-submissions-software-contained-medical-devices. ↩
-
ISO 14971 Medical devices — Application of risk management to medical devices. ↩
-
ISO 14971 Medical devices — Application of risk management to medical devices. ↩
-
ISO 14971 Medical devices – Application of risk management to medical devices. ↩
-
Available at https://www.fda.gov/inspections-compliance-enforcement-and-criminalinvestigations/inspection-guides/glossary-computer-system-software-development-terminology-895. ↩
-
Available at https://www.fda.gov/regulatory-information/search-fda-guidance-documents/guidance-contentpremarket-submissions-software-contained-medical-devices. ↩
-
Haddon W, Baker SP. “Injury Control.” Preventative and Community Medicine. Eds. Clark D, MacMahon B. New York: Little Brown & Company, 1981. 109-140. Abs: A readable discussion of basic injury reduction strategies from some of the most experienced in the field. ↩
-
Available at https://www.fda.gov/regulatory-information/search-fda-guidance-documents/deciding-whensubmit-510k-change-existing-device. ↩
-
Available at https://www.fda.gov/regulatory-information/search-fda-guidance-documents/deciding-whensubmit-510k-software-change-existing-device. ↩
-
Available at https://www.fda.gov/regulatory-information/search-fda-guidance-documents/deciding-whensubmit-510k-change-existing-device. ↩
-
Available at https://www.fda.gov/regulatory-information/search-fda-guidance-documents/deciding-whensubmit-510k-software-change-existing-device. ↩
-
See 21 CFR 812. ↩
-
Available at https://www.fda.gov/regulatory-information/search-fda-guidance-documents/significant-riskand-nonsignificant-risk-medical-device-studies. ↩
-
See 21 CFR 860.7. ↩
-
Available at https://www.fda.gov/regulatory-information/search-fda-guidance-documents/device-labelingguidance-g91-1-blue-book-memo. ↩
-
Available at https://www.fda.gov/regulatory-information/search-fda-guidance-documents/labeling-regulatoryrequirements-medical-devices-fda-89-4203. ↩
