
About this Transcript 🔗
This document is a transcript of an official FDA (or IMDRF) guidance document. We transcribe the official PDFs into HTML so that we can share links to particular sections of the guidance when communicating internally and with our clients. We do our best to be accurate and have a thorough review process, but occasionally mistakes slip through. If you notice a typo, please email a screenshot of it to Miljana Antic at mantic@innolitics.com so we can fix it.
Preamble 🔗
Document issued on: March 13, 2007
The draft of this document was issued on March 12, 2003.
For questions regarding this document, contact Kristen Meier at 240-276-3060, or send an e-mail to kristen.meier@fda.hhs.gov.
Contains non-binding guidance.
1. Background 🔗
This guidance is intended to describe some statistically appropriate practices for reporting results from different studies evaluating diagnostic tests and identify some common inappropriate practices. The recommendations in this guidance pertain to diagnostic tests where the final result is qualitative (even if the underlying measurement is quantitative). We focus special attention on the practice called discrepant resolution and its associated problems.
On February 11, 1998, the Center for Devices and Radiological Health convened a joint meeting of the Microbiology, Hematology/Pathology, Clinical Chemistry/Toxicology, and Immunology Devices Panels. The purpose of the meeting was to obtain recommendations on “appropriate data collection, analysis, and resolution of discrepant results, using sound scientific and statistical analysis to support indications for use of the in vitro diagnostic devices when the new device is compared to another device, a recognized reference method or ‘gold standard,’ or other procedures not commonly used, and/or clinical criteria for diagnosis.” Using the input from that meeting, a draft guidance document was developed discussing some statistically valid approaches to reporting results from evaluation studies for new diagnostic devices. The draft guidance was released for public comment March 12, 2003.
Following publication of the draft guidance, FDA received 11 comments. Overall, the comments were favorable and requested additional information be included in the final guidance. Some respondents requested greater attention to the use of standard terminology.
Correct use of terminology for describing performance results is important to ensure safe and effective use of a diagnostic device. Whenever possible, this guidance uses internationally accepted terminology and definitions as compiled in the Clinical and Laboratory Standards Institute (CLSI) Harmonized Terminology Database.1 This guidance also uses terms as they are defined in the STARD (Standards for Reporting of Diagnostic Accuracy) Initiative.2 The STARD Initiative pertains to studies of diagnostic accuracy. While the STARD Initiative does not specifically address studies designed to demonstrate diagnostic device equivalence, many of the reporting concepts are still applicable.
FDA’s guidance documents, including this guidance, do not establish legally enforceable responsibilities. Instead, guidances describe the Agency’s current thinking on a topic and should be viewed only as recommendations, unless specific regulatory or statutory requirements are cited. The use of the word should in Agency guidances means that something is suggested or recommended, but not required.
We believe we should consider the least burdensome approach in all areas of medical device regulation. This guidance reflects our careful review of the relevant scientific and legal requirements and what we believe is the least burdensome way for you to comply with those requirements. However, if you believe that an alternative approach would be less burdensome, please contact us so we can consider your point of view. You may send your written comments to the contact person listed in the preface to this guidance or to the CDRH Ombudsman. Comprehensive information on CDRH’s Ombudsman, including ways to contact him, can be found on the Internet at http://www.fda.gov/cdrh/ombudsman/.
2. Scope 🔗
This document provides guidance for the submission of premarket notification (510(k)) and premarket approval (PMA) applications for diagnostic devices (tests). This guidance addresses the reporting of results from different types of studies evaluating diagnostic devices with two possible outcomes (positive or negative) in PMAs and 510(k)s. The guidance is intended for both statisticians and non-statisticians.
This guidance does not address the fundamental statistical issues associated with design and monitoring of clinical studies for diagnostic devices.
3. Introduction 🔗
This section provides an explanation of the concepts relevant to this guidance. We note at the outset that evaluation of a new diagnostic test should compare a new product’s outcome (test results) to an appropriate and relevant diagnostic benchmark using subjects/patients from the intended use population; that is, those subjects/patients for whom the test is intended to be used. In STARD, this is called the target population.
Other important concepts and definitions include the following:
Types of test results
The method of comparison depends on the nature of the test results. Diagnostic test results (outcomes) are usually classified as either quantitative or qualitative. A quantitative result is a numerical amount or level, while a qualitative result usually consists of one of only two possible responses; for example, diseased or non-diseased, positive or negative, yes or no. This document pertains to diagnostic tests where the final result is qualitative (even if the underlying measurement is quantitative). Quantitative tests and tests with ordinal outcomes (more than two possible outcomes, but ordered) are not discussed here.
We also assume throughout that your study data do not include multiple samples from single patients.
Purpose of a qualitative diagnostic test
A qualitative diagnostic test (test) is designed to determine whether a target condition is present or absent in a subject from the intended use population. As defined in STARD, the target condition (condition of interest) “can refer to a particular disease, a disease stage, health status, or any other identifiable condition within a patient, such as staging a disease already known to be present, or a health condition that should prompt clinical action, such as the initiation, modification or termination of treatment.”
FDA recommends your labeling characterize diagnostic test performance for use by all intended users (laboratories, health care providers, and/or home users).
Benchmarks
FDA recognizes two major categories of benchmarks for assessing diagnostic performance of new qualitative diagnostic tests. These categories are (1) comparison to a reference standard (defined below), or (2) comparison to a method or predicate other than a reference standard (non-reference standard). The choice of comparative method will determine which performance measures may be reported in the label.
Diagnostic accuracy and the reference standard
The diagnostic accuracy of a new test refers to the extent of agreement between the outcome of the new test and the reference standard. We use the term reference standard as defined in STARD. That is, a reference standard is “considered to be the best available method for establishing the presence or absence of the target condition.” It divides the intended use population into only two groups (condition present or absent) and does not consider the outcome of the new test under evaluation.
The reference standard can be a single test or method, or a combination of methods and techniques, including clinical follow-up. If a reference standard is a combination of methods, the algorithm specifying how the different results are combined to make a final positive/negative classification (which may include the choice and ordering of these methods) is part of the standard. Examples of reference standards include the diagnosis of myocardial infarction using the WHO (World Health Organization) standards, the diagnosis of lupus or rheumatoid arthritis using American Rheumatology guidelines, or the diagnosis of H. pylori infections by use of combinations of culture, histology, and urease testing.
The determination of what constitutes the “best available method” and whether that method should be considered a “reference standard” is established by opinion and practice within the medical, laboratory, and regulatory community.
Sometimes there are several possible methods that could be considered. Sometimes no consensus reference standard exists. Or, a reference standard may exist, but for a non-negligible percentage of the intended use population, the reference standard is known to be in error. In all these situations, we recommend you consult with FDA on your choice of reference standard before you begin your study.
We point out that some definitions of diagnostic accuracy (see CLSI harmonized terminology database) require that the reference standard and target condition refer only to a well-defined clinical disorder. The definitions used in this document are broader. For example, the target condition could be a well-defined health condition or a condition that prompts a clinical action such as the initiation of a treatment.
Measures that describe diagnostic accuracy
There are different ways to describe diagnostic accuracy. Appropriate measures include estimates of sensitivity and specificity pairs, likelihood ratio of positive and negative result pairs, and ROC (Receiver Operating Characteristic) analysis along with confidence intervals. Refer to the most current edition of CLSI Approved Guidelines EP12-A and GP10-A; the texts by Lang and Secic (1997), Pepe (2003), Zhou et al. (2002); the references within these texts; and the bibliography at the end of this document. To help interpret these measures, we recommend you provide the definition of condition of interest, the reference standard, the intended use population, and a description of the study population.
Sensitivity and specificity
In studies of diagnostic accuracy, the sensitivity of the new test is estimated as the proportion of subjects with the target condition in whom the test is positive. Similarly, the specificity of the test is estimated as the proportion of subjects without the target condition in whom the test is negative (see the Appendix for an example of this calculation). These are only estimates for sensitivity and specificity because they are based on only a subset of subjects from the intended use population; if another subset of subjects were tested (or even the same subjects tested at a different time), then the estimates of sensitivity and specificity would probably be numerically different. Confidence intervals and significance levels quantify the statistical uncertainty in these estimates due to the subject/sample selection process. This type of uncertainty decreases as the number of subjects in the study increases.
Positive and negative predictive value
You may also compute other quantities to help characterize diagnostic accuracy. These methods include the predictive value of a positive result (sometimes called positive predictive value or PPV) and predictive value of a negative result (sometimes called negative predictive value or NPV) pair. These quantities provide useful insight into how to interpret test results. You may refer to the extensive literature on how to calculate and interpret these measures. (See most current edition of CLSI EP12-A, Lang and Secic (1997), Pepe (2003), Zhou et al. (2002), the references within the texts, and the bibliography at the end of this document.) Further discussion of these measures is beyond the scope of this document.
Bias
Sensitivity and specificity estimates (and other estimates of diagnostic performance) can be subject to bias. Biased estimates are systematically too high or too low. Biased sensitivity and specificity estimates will not equal the true sensitivity and specificity, on average. Often the existence, size (magnitude), and direction of the bias cannot be determined. Bias creates inaccurate estimates.
FDA believes it is important to understand the potential sources of bias to avoid or minimize them. Simply increasing the overall number of subjects in the study will do nothing to reduce bias. Alternatively, selecting the “right” subjects, changing study conduct, or data analysis procedures may remove or reduce bias.
Two sources of bias that originally motivated the development of this guidance include error in the reference standard and incorporation of results from the test under evaluation to establish the target condition. This guidance discusses problems arising from these and other sources of bias and describes how to minimize these problems in your study design and data analysis. This guidance does not attempt to discuss all possible sources of bias and how to avoid them. For comprehensive discussions on bias and diagnostic device studies, see Begg (1987), Pepe (2003), Zhou et al. (2002), and the references cited in these texts.
When a non-reference standard is used for comparison
When a new test is evaluated by comparison to a non-reference standard, sensitivity and specificity are not appropriate terms to describe the comparative results. Information on the accuracy or “correctness” of the new test cannot be estimated directly. Instead, when a non-reference standard is used for comparison, FDA recommends you demonstrate the ability of the candidate test to agree sufficiently with the comparative method or predicate. A question addressed in this document is how to report results from a study evaluating a new diagnostic test when the comparative method is not a reference standard.
4. Benchmark and Study Population Recommendations 🔗
FDA recommends you carefully plan your study before collecting the first specimen or taking the first measurement. This includes determining whether you want to report diagnostic accuracy or device agreement. If you want to report diagnostic accuracy, FDA recommends your evaluation include the use of a reference standard on at least some of the subjects.
We recommend you contact CDRH early to discuss possible study designs and statistical analyses prior to any data collection for the clinical study.3 Often there are promising advanced statistical methods that may be appropriate, and new statistical analysis techniques are constantly being developed. The list of references at the end of this document includes a variety of approaches. Discussing your planned study with CDRH before starting may save time and money.
4.1 Comparisons with the Benchmark 🔗
The choice of comparative benchmark and the methods of comparison and reporting are influenced by the existence and/or practical applicability of a reference standard. Depending on the availability of a reference standard, FDA makes the following recommendations regarding the choice of comparative benchmark:
- If a reference standard is available: use it to estimate sensitivity and specificity
- If a reference standard is available, but impractical: use it to the extent possible. Calculate estimates of sensitivity and specificity adjusted to correct for any (verification) bias that may have been introduced by not using the reference standard to its fullest extent.
- If a reference standard is not available or unacceptable for your particular intended use and/or intended use population: consider whether one can be constructed. If so, calculate estimated sensitivity and specificity under the constructed standard.
- If a reference standard is not available and cannot be constructed: calculate and report measures of agreement (see Appendices).
We now provide more details on these recommendations:
If a reference standard is available
From a purely statistical perspective, FDA believes that the best approach is to designate a reference standard and compare the new test to the designated reference standard, drawing from subjects who are representative of the intended use population. We recommend you consult with FDA prior to planning a study to ensure the designated reference standard will meet Agency needs. In this situation, sensitivity and specificity have meaning, and you can easily calculate the estimates. The Appendices contain a numerical example.
If a reference standard is available, but impractical
If you determine that using a reference standard on all subjects is impractical or not feasible, FDA recommends you obtain estimates of sensitivity and specificity using the new test and a comparative method (other than a reference standard) on all subjects, and use the reference standard on just a subset of subjects (sometimes called partial verification studies or two-stage studies).
For example, if you apply the designated reference standard to a random subset of all subjects, or to all subjects where the new test and the comparative method disagree and to a random sample of subjects where they agree, then it is possible to compute adjusted estimates (and variances) of sensitivity and specificity. In this case FDA recommends you retest a sufficient number of subjects to estimate sensitivity and specificity with reasonable precision.
Note that the simple formulas for calculating sensitivity and specificity described in the Appendix are not correct for this design and such naive calculations would give biased estimates of sensitivity and specificity. This type of bias is an example of verification or work-up bias. For details see Begg (1987), Pepe (2003), or Zhou et al. (2002).
Determining how large a subset to choose, the particular subset to choose, and how to calculate the performance measures is currently an area of active statistical research. See Albert (2006), Albert & Dodd (2004, 2006), Hawkins et al. (2001), Kondratovich (2003), Pepe (2003), Zhou et al. (2002), and references cited within these references. Since this approach can be statistically complicated, FDA recommends you consult with a CDRH statistician before using this approach.
In rare circumstances, it may be possible to estimate sensitivity and specificity without using a reference standard in the study. This may be reasonable, for example, when the sensitivity and specificity of the designated comparative method are well established from previous evaluations against a reference standard in similar subject populations. Further elaboration of this subject is beyond the scope of this document. Here too, FDA recommends you consult with a CDRH statistician before using this approach.
If a reference standard is not available, but might be constructed An expert panel (FDA advisory panel or other panel) may be able to develop a set of clinical criteria (or a combination of reference tests and confirmatory clinical information) that would serve as a designated reference standard. While this approach may be more time-consuming up front, if successful, you can easily calculate estimates of sensitivity and specificity. In this situation, FDA recommends
- the test label clearly describe the designated reference standard that was constructed
- the new reference standard be created independently from the analysis of results of the new diagnostic test (ideally, in advance of collecting any specimens)
- you consult with CDRH medical officers and statisticians prior to constructing a reference standard.
If a reference standard is not available and cannot be constructed When a new test is evaluated by comparison to a non-reference standard, you cannot directly calculate unbiased estimates of sensitivity and specificity. Therefore, the terms sensitivity and specificity are not appropriate to describe the comparative results. Instead, the same numerical calculations are made, but the estimates are called positive percent agreement and negative percent agreement, rather than sensitivity and specificity. This reflects that the estimates are not of accuracy but of agreement of the new test with the non-reference standard.
In addition, quantities such as positive predictive value, negative predictive value, and the positive and negative likelihood ratios cannot be computed since the subjects’ condition status (as determined by a reference standard) is unknown.
In this situation, FDA recommends you report
- the 2x2 table of results comparing the candidate test with the comparative method
- a description of the comparative method and how it was performed
- the pair of agreement measures along with their confidence
The Appendices provide a numerical example.
We adopt the terms “positive percent agreement” and “negative percent agreement” with the following cautionary note. Agreement of a new test with the non-reference standard is numerically different from agreement of the non-reference standard with the new test (contrary to what the term “agreement” implies). Therefore, when using these measures of agreement, FDA recommends you clearly state the calculations being performed.
One major disadvantage with agreement measures is that agreement is not a measure of “correctness.” Two tests could agree and both be wrong. In fact, two tests could agree well, but both have poor sensitivity and specificity. However, when two tests disagree, that does not mean that the new test is wrong and the comparative method is right.
One should also be aware that measures of overall agreement (including both overall percent agreement and Cohen’s Kappa) can be misleading in this setting. In some situations, overall agreement can be good when either positive or negative percent agreement is very low. For this reason, FDA discourages the stand-alone use of measures of overall agreement to characterize the diagnostic performance of a test.
There has been much statistical research on how to estimate diagnostic accuracy of a new test when a reference standard is not available or does not exist. Albert and Dodd (2004), Pepe (2003), and Zhou et al. (2002) provide reviews of some of this research, which includes use of latent class models and Bayesian models. These model-based approaches can be problematic for the purpose of estimating sensitivity and specificity because it is often difficult to verify that the model and assumptions used are correct. More troublesome is that different models can fit the data equally well, yet produce very different estimates of sensitivity and specificity. For these types of analyses, FDA recommends reporting a range of results for a variety of models and assumptions. FDA also recommends you consult with a CDRH statistician before using these approaches.
4.2 Selecting the Study Population 🔗
In addition to choosing an appropriate comparative benchmark, evaluating a new test also involves choosing an appropriate set of:
- subjects or specimens to be tested
- individuals and laboratories to perform the tests
- conditions under which the tests will be conducted.
Spectrum bias
Estimates of diagnostic accuracy are subject to spectrum bias when the subjects included in the study do not include the complete spectrum of patient characteristics; that is, important patient subgroups are missing. See Begg (1987), Pepe (2003), or Zhou et al. (2002). For example, there are studies that include only very healthy subjects and subjects with severe disease, omitting the intermediate and typically more difficult cases to diagnose. The accuracy measures reported from these studies are subject to spectrum bias.
Eliminating the difficult cases produces an overly optimistic picture of how the device performs in actual use. Therefore, FDA recommends the set of subjects and specimens to be tested include:
- subjects/specimens across the entire range of disease states
- subjects/specimens with relevant confounding medical conditions
- subjects/specimens across different demographic groups.
If the set of subjects and specimens to be evaluated in the study is not sufficiently representative of the intended use population, the estimates of diagnostic accuracy can be biased.
External validity
A study has high external validity if the results from the study are sufficiently reflective of the “real world” performance of the device in the intended use population. Selection of the appropriate set of subjects and/or specimens is not in itself sufficient to ensure high external validity. Although detailed discussion of external validity is beyond the scope of this document, FDA generally recommends:
- using the final version of the device according to the final instructions for use
- using several of these devices in your study
- including multiple users with relevant training and range of expertise
- covering a range of expected use and operating conditions.
See Rothwell (2006) for a non-technical discussion in the context of randomized trials.
5. Reporting Recommendations 🔗
Similar reporting principles apply to any study evaluating a diagnostic test, regardless of whether the comparative benchmark is a reference standard.
Reporting the context of the study
Performance measures should be interpreted in the context of the study population and study design. Sensitivity and specificity cannot be interpreted by themselves; additional information is needed. For example, estimated sensitivity and specificity of the same test can differ from study to study, depending on the types of subjects included in the study and whether an obsolete reference standard is used versus a reference standard currently accepted by the clinical community today.
Before presenting results, FDA recommends you describe or define the:
- intended use population
- study population
- condition of interest (precise definition of condition explaining how those subjects with the condition of interest are distinguished from those without)
- designated comparative benchmark (reference standard or comparative method).
FDA also recommends you discuss:
- the rationale for the choice of designated comparative benchmark
- the strengths and limitations likely to result from selection of that benchmark.
Defining the conditions of use
FDA recommends you define the conditions of use under which the candidate test and the reference standard or comparative method are performed. These may include:
- operator experience
- clinical laboratory facility or other test setting
- controls applied
- specimen acceptance criteria.
Descriptions of comparative results and methods
FDA recommends you include in your results a clear description of all methods used and how and what data were collected, such as:
- subject recruitment procedures
- subject demographics
- subject and specimen inclusion and exclusion criteria
- specimen collection procedures
- time of specimen collection and testing
- types of specimens collected
- number of specimens collected and tested and number discarded
- number of specimens included in final data analysis
- specimen collection devices (if applicable)
- specimen storage and handling procedures.
Reporting study results
FDA recommends you report all results by
- clinical site or specimen collection site,
- specimen testing or processing site, and
- relevant clinical and demographic subgroups.
Tabular comparisons
FDA recommends you report tabular comparisons of the candidate test outcome to the reference standard or comparative method. (For example, we recommend you report the 2x2 table of results such as those in the Appendix.)
Measures of accuracy
FDA recommends you report measures of diagnostic accuracy (sensitivity and specificity pairs, positive and negative likelihood ratio pairs) or measures of agreement (percent positive agreement and percent negative agreement) and their two-sided 95 percent confidence intervals. We recommend reporting these measures both as fractions (e.g., 490/500) and as percentages (e.g., 98.0%). The Appendices contain a numerical example.
Underlying quantitative result
For qualitative tests derived from an underlying quantitative result, FDA recommends you provide descriptive summaries that include:
- ranges of results
- histograms of results by condition status (if known)
- Receiver Operating Characteristic (ROC) Plots (if condition status is known).
The CLSI document GP10 Assessment of the Clinical Accuracy of Laboratory Tests Using Receiver Operating Characteristic (ROC) Plots provides further guidance on this topic.
Accounting of subjects and test results
FDA recommends you provide a complete accounting of all subjects and test results, including:
- number of subjects planned to be tested
- number tested
- number used in final analysis
- number omitted from final analysis.
Other results
FDA recommends you provide the number of ambiguous4 results for candidate tests, stratified by reference standard outcome or comparative outcome.
Reporting intended use population results separately
FDA recommends you report results for those subjects in the intended use population separately from other results. It may be useful to report comparative results for subjects who are not part of the intended use population, but we recommend they not be pooled together. For example, if healthy individuals are not part of the intended use population, we recommend those results be reported separately from results for the intended use population. Results from patients outside the intended use population should not be labeled as “specificity.” The term specificity is appropriate to describe how often a test is negative only in subjects from the intended use population for whom the target condition is absent.
Rare condition of interest
When the condition of interest is rare, studies are sometimes enriched with reference standard positive subjects, potentially making the results inappropriate for pooling with other positive results. We recommend you consult with FDA on this issue.
Archived collections
If your test is evaluated using specimens retrospectively obtained from archived collections, sensitivity and specificity claims may or may not be appropriate. These claims may be appropriate if the archived specimens are representative of specimens from subjects in the intended use population, with and without the target condition, including unclear cases. FDA recommends you provide a description of the results, indicating:
- the nature of the specimens studied
- how the target condition status was determined
- the limitations introduced through selective sampling.
6. Statistically Inappropriate Practices 🔗
Some common practices for reporting results are statistically inappropriate because they are misleading or can lead to inaccurate estimates of test performance. These practices most often arise when a new test is compared to a comparative method other than a reference standard.
Comparing a new test to a non-reference standard does not yield true performance. If the new test is better than the non-reference standard, the agreement will be poor. Alternatively, the agreement could be poor because the non-reference standard is fairly accurate and the new test is inaccurate. There is no statistical solution to determining which scenario is the true situation.
When comparing a new test to a non-reference standard, FDA makes the following recommendations regarding four common practices that we believe give misleading or incorrect results.
-
Avoid use of the terms “sensitivity” and “specificity” to describe the comparison of a new test to a non-reference standard When a new test is evaluated by comparison to a non-reference standard, it is impossible to calculate unbiased estimates of sensitivity and specificity. In addition, quantities such as positive predictive value, negative predictive value, and the positive and negative likelihood ratios cannot be computed since the subjects’ condition status (as determined by a reference standard) is unknown.
For this reason, FDA recommends you report
- the 2x2 table of results comparing the new test with the non-reference standard
- a description of the non-reference standard
- measures of agreement and corresponding confidence intervals.
FDA recommends the use of the terms positive percent agreement and negative percent agreement with the non-reference standard to describe these results. Agreement measures are discussed in more detail in the Appendices.
-
Avoid elimination of equivocal5 results If a test can (per the test instructions) produce a result which is anything other than positive or negative then it is not technically a qualitative test (since more than two outcomes are possible). In that case the measures described in this guidance do not directly apply. Discarding or ignoring these results and performing the calculations in this guidance will likely result in biased performance estimates.
To address this issue, one option is to report two different sets of performance measures
- one set of measures based on including the equivocal results with the test positive results
- a second set of measures based on including the equivocal results with the test negative results.
This may or may not be reasonable for your situation. FDA recommends you consult with FDA statisticians on how to handle these types of results.
-
Avoid the use of outcomes altered or updated by discrepant resolution You should not use outcomes that are altered or updated by discrepant resolution to estimate the sensitivity and specificity of a new test or agreement between a new test and a non-reference standard. When a new test is evaluated by comparison to a non-reference standard, discrepancies (disagreement) between the two methods may arise because of errors in the test method or errors in the non-reference standard. Since the non- reference standard may be wrong, calculations of sensitivity and specificity based on the non-reference standard are statistically biased. A practice called discrepant resolution has been suggested to get around the bias problem.
As the name implies, discrepant resolution focuses on subjects where there is a discrepancy; that is, where the new test and the non-reference standard disagree. In the simplest situation, discrepant resolution can be described as a two-stage testing process:
- Stage 1: Testing all subjects using the new test and the non-reference standard
- Stage 2: When the new test and non-reference standard disagree, using a resolver (a reference standard or a second non-reference standard) to see which one is “right.”
A numerical example describing discrepant resolution appears in the Appendix. If the resolver is a reference standard, this process provides the condition status for the subjects re-tested with the resolver, but it does not provide the condition status for subjects when the new test agrees with the non-reference standard (usually most of the subjects). Even when the new test and non-reference standard agree, they may both be wrong.
FDA does not recommend the process used by some investigators whereby the resolver is used to revise the original 2x2 table of results (new test versus non- reference standard). We believe the original 2x2 table is inappropriately “revised” in this method because:
- when the original two results agree, you assume (without supporting evidence) that they are both correct and do not make any changes to the table
- when the original results disagree, and the non-reference standard disagrees with the resolver, you reclassify (change) the non-reference standard result to the resolver result.
The revised 2x2 table based on discrepant resolution is misleading because the columns are not clearly defined and do not necessarily represent condition status, as assumed. The assumption that results that agree are correct is not tested and may be far from valid. FDA recommends you do not present such a table in your final analysis because it may be very misleading. Because the calculations of sensitivity and specificity from such a revised 2x2 table are not valid estimates of performance, they should not be reported.
FDA is not aware of any scientifically valid ways to estimate sensitivity and specificity by resolving only the discrepant results, even when the resolver is a reference standard. To obtain unbiased estimates of sensitivity and specificity, FDA believes
- the resolver must be a reference standard, and
- you must resolve at least a subset of the concordant subjects.
Discrepant resolution with a reference standard resolver can tell you whether the new test or the non-reference standard is right more of the time, but you cannot quantify how much more. If the resolver is not a reference standard, the resolver test results can provide little or no useable information about the performance of the new test. Resolving discrepancies using repeat testing by the new test or the non-reference standard also does not provide any useful information about performance.
-
Avoid comparison of the results of a new test to the outcome of a testing algorithm that combines several comparative methods (non-reference standards), if the algorithm uses the outcome of the new test When evaluating some types of tests, the comparative “procedure” is not a single test, but the outcome of a combination of several comparative methods and possibly clinical information. Often, two or more comparative methods are performed and interpreted according to a pre-specified testing sequence or algorithm to determine condition status.
The decision to use a second or third comparative method may depend on the outcome of the initial comparative method. This approach may be statistically reasonable. However, FDA believes this approach is not valid if the algorithm uses the outcome of the new unproven test. For example, the decision to use an additional comparative method should not be based on whether the new test is positive or negative.
FDA believes it is potentially misleading to establish the performance of a new test by comparing it to a procedure that incorporates the same new test. Any non-reference standard created in this manner will likely be biased in favor of the new test; that is, it will tend to produce overestimates of agreement of the new test with the non-reference standard.
Summary
In summary, when reporting results from a study evaluating a diagnostic test, FDA believes it is inappropriate to:
- use the terms “sensitivity” and “specificity” to describe the comparison of a new test to a non-reference standard
- discard equivocal new test results when calculating measures of diagnostic accuracy or agreement
- use outcomes that are altered or updated by discrepant resolution to estimate the sensitivity and specificity of a new test or agreement between a new test and a non-reference standard
- compare the results of a new test to the outcome of a testing algorithm that combines several comparative methods (non-reference standards), if the algorithm uses the outcome of the new test.
7. Appendices 🔗
7.1 Calculating Estimates of Sensitivity and Specificity 🔗
Sensitivity and specificity are basic measures of performance for a diagnostic test. Together, they describe how well a test can determine whether a specific condition is present or absent. They each provide distinct and equally important information, and FDA recommends they be presented together:
- Sensitivity refers to how often the test is positive when the condition of interest is present
- Specificity refers to how often the test is negative when the condition of interest is absent.
Note that a diagnostic test where sensitivity equals [1– specificity] has no diagnostic value. That is, if the percent of subjects with positive test results when the condition is present (sensitivity) is the same as the percent of subjects with positive test results when the condition is absent (1– specificity), then the new test outcome is unaffected by the condition of interest, and it has no diagnostic value for that condition of interest. However, a test where both sensitivity and specificity are close to 1 has good diagnostic ability.
Usually, to estimate sensitivity and specificity, the outcome of the new test is compared to the reference standard using subjects who are representative of the intended use (both condition present and condition absent) population.
We assume throughout that your study data do not include multiple samples from single patients. If you do have such data, we recommend that you consult with FDA statisticians on the appropriate calculation methods.
Results are typically reported in a 2x2 table such as Table 1.
Table 1. Common 2x2 table format for reporting results comparing a new test outcome to the reference standard outcome
The new test has two possible outcomes, positive (+) or negative (−). Subjects with the condition of interest are indicated as reference standard (+), and subjects without the condition of interest are indicated as reference standard (−).

Where TP = number of true positive events
FP = number of false positive events
TN = number of true negative events
FN = number of false negative events
From Table 1, estimated sensitivity is the proportion of subjects with the condition of interest (reference standard+) that are New Test+. Estimated specificity is the proportion of subjects without the condition of interest (reference standard−) that are New Test−. The formulas are as follows.
estimated sensitivity = 100% x TP/(TP+FN)
estimated specificity = 100% x TN/(FP +TN)
Here is an example of this calculation. Suppose one specimen is taken from each of 220 subjects in the intended use population. Each specimen is tested by the new test and the reference standard. Fifty-one (51) subjects have the condition of interest and 169 do not. The results are presented in a 2x2 table format in Table 2.
Table 2. Example of results comparing a new test to reference standard for 220 subjects
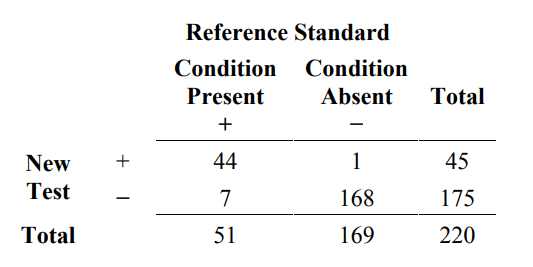
From Table 2, estimated sensitivity and specificity are calculated in the following manner:
estimated sensitivity = 100% x 44/51 = 86.3%
estimated specificity = 100% x 168/169 = 99.4%
Two-sided 95% score confidence intervals for sensitivity and specificity are (74.3%, 93.2%) and (96.7%, 99.9%), respectively. See Altman et al. (2000) and the most current edition of CLSI EP12-A for a brief discussion about computing score confidence intervals, and alternatively, exact (Clopper-Pearson) confidence intervals for sensitivity and specificity.
7.2 Calculating an Estimate of Agreement 🔗
When a new test is compared to a non-reference standard rather than to a reference standard, the usual sensitivity and specificity type calculations from the 2x2 table will produce biased estimates of sensitivity and specificity because the non-reference standard is not always correct. In addition, quantities such as positive predictive value, negative predictive value, and the positive and negative likelihood ratios cannot be computed since the subjects’ condition status (as determined by a reference standard) is unknown.
However, being able to describe how often a new test agrees with a non-reference standard may be useful.
To do this, a group of subjects (or specimens from subjects) is tested twice, once with the new test and once with the non-reference standard. The results are compared and may be reported in a 2x2 table such as Table 3.
Table 3. Common 2x2 table format for reporting results comparing a new test to a non-reference standard

The difference between Table 3 and Table 1 is that the columns of Table 3 do not represent whether the subject has the condition of interest as determined by the reference standard. The entries in Table 3 (a, b, c, d) no longer represent (TP, FP, FN, TN), respectively. Therefore, data from Table 3 cannot be interpreted in the same way as Table 1. Data from Table 1 provides information on how often the new test is correct; whereas, data from Table 3 provides information on how often the new test agrees with a non-reference standard.
From Table 3, you can compute several different statistical measures of agreement. A discussion by M.M. Shoukri on different types of agreement measures appears under “Agreement, Measurement of” in the Encyclopedia of Biostatistics (1998). Two commonly used measures are the overall percent agreement and (Cohen’s) kappa. The simplest measure is overall percent agreement: the percentage of total subjects where the new test and the non-reference standard agree. You can calculate estimated overall percent agreement from Table 3 in the following way:
overall percent agreement = 100% x (a+d)/(a+b+c+d)
Overall percent agreement does not by itself adequately characterize the agreement of a test with the non-reference standard. Two different 2x2 tables can have the same overall agreement with the same value for (b+c) but have very different values for b and c individually. Therefore, it is more useful to report a pair of agreement measures, positive percent agreement (PPA) and negative percent agreement (NPA):
positive percent agreement = 100% x a/(a+c)
negative percent agreement = 100% x d/(b+d)
The overall agreement will always lie somewhere between the positive percent agreement and the negative percent agreement.
We adopt the terms “positive percent agreement” and “negative percent agreement” with the following cautionary note. Agreement of a new test with the non-reference standard is numerically different from agreement of the non-reference standard with the new test (contrary to what the term “agreement” implies). As defined here, the positive percent agreement is the proportion of non-reference standard+ subjects that are New Test+ (analogous to a sensitivity calculation).
One could also compute the proportion of New Test+ subjects that are non-reference standard+ and get a different number. Therefore, when calculating positive and negative percent agreement, FDA recommends explicitly stating the calculation being performed.
As an example of some of these calculations, consider the same 220 subjects as before. After all 220 are tested with both the new test and the non-reference standard, we have the following results.
Table 4. Example of results comparing a new test to a non-reference standard for 220 subjects

From Table 4, calculate the agreement measures as follows:
positive percent agreement (new / non ref. std.) = 100% x 40/44 = 90.9%
negative percent agreement (new / non ref. std.)= 100% x 171/176 = 97.2%
overall percent agreement = 100% x (40+171)/220 = 95.9%
Two-sided 95% score confidence intervals for positive percent agreement and negative percent agreement conditional on the observed non-reference standard results (ignoring variability in the non-reference standard) are (78.8%, 96.4%) and ( 93.5%, 98.8%), respectively. A two-sided 95% score confidence interval for overall percent agreement is (92.4%, 97.8%). See Altman et al. (2000) and the most current edition of CLSI EP12-A for a brief discussion about computing score confidence intervals and alternatively, how to calculate exact (Clopper-Pearson) confidence intervals.
To illustrate how measures of overall agreement can be misleading, suppose that 572 subjects are tested with two new tests (New Test A and New Test B) and the non- reference standard. The comparative results are presented in Tables 5A and 5B. Table 5A is an example of how overall agreement can be high, but positive percent agreement is low. The overall agreement is 96.5% (532/572), but the positive percent agreement (new/non ref. std.) is only 67.8% (40/59).
Tables 5A and 5B together are an example of how overall agreement remains the same, but the performance of the two tests is different, as evidenced by the different positive and negative percent agreement results. For both tests, the overall agreement is 96.5% (552/572). For New test A, the positive and negative percent agreement (new/non ref. std.) results are 67.8% (40/59) and 99.8% (512/513), respectively. The corresponding results for New Test B are different: the results are 97.6% (40/41) and 96.4% (512/531), respectively.
Table 5A. Example of results comparing new test A to a non-reference standard for 572 subjects where the positive percent agreement is low, but overall agreement is high

positive percent agreement (new / non ref. std.) = 100% x 40/59 = 67.8%
negative percent agreement (new / non ref. std.) = 100% x 512/513 = 99.8%
overall percent agreement = 100% x (40+512)/572 = 96.5%
Table 5B. Example of results comparing new test B to a non-reference standard for 572 subjects where the positive percent agreement is high, and overall agreement is high

positive percent agreement (new / non ref. std.) = 100% x 40/41 = 97.6%
negative percent agreement (new / non ref. std.) = 100% x 512/531 = 96.4%
overall percent agreement = 100% x (40+512)/572 = 96.5%
Thus, measures of overall agreement are not in themselves a sufficient characterization of the performance of a test.
All agreement measures, including kappa, overall agreement, positive percent agreement, and negative percent agreement have two major disadvantages:
- “Agreement” does not mean “correct.”
- Agreement changes depending on the prevalence (the relative frequency of the condition of interest in a specified group; also called pre-test probability).
We now explore these disadvantages.
“Agreement” does not mean “correct”
Note from the examples in Tables 2 and 4 that the non-reference standard did not correctly classify all 220 subjects. The non-reference standard classified 44 subjects as positive and 176 as negative (from Table 4). From Table 2, in truth, 51 subjects have the condition of interest, and 169 do not. Since the non-reference standard is wrong sometimes, you cannot calculate unbiased estimates of sensitivity and specificity from Table 4; instead, you can calculate agreement.
When two tests agree, one cannot assume they are also correct. In order to demonstrate this, we need a three-way comparison between the new test result, the non-reference standard result, and the reference standard. A useful way to present the three-way comparison is shown in Table 6A.
Table 6A. A three-way presentation of results comparing the new test, the non- reference standard, and the reference standard
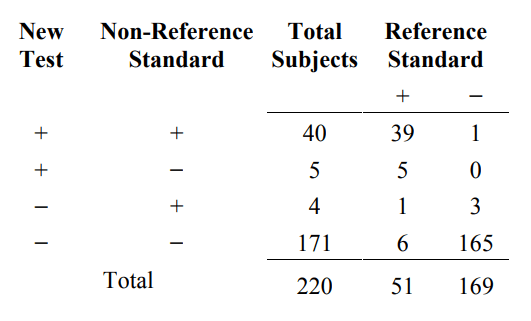
From the first and fourth rows of Table 6A, the new test and the non-reference standard agree for 40+171=211 subjects, but they agree and are both wrong for 6+1=7 subjects.
As an alternate to the 4x2 Table 6A, one may choose to present these results as two separate 2x2 tables, partitioned by the reference standard (Table 6B). The data are the same, but sometimes the presentation in Table 6B provides different insight from the presentation in Table 6A.
Table 6B. An alternative presentation of results comparing the new test, the non- reference standard, and the reference standard

Agreement changes depending on prevalence
Regarding the second disadvantage, the agreement between two methods is usually different in subjects with the condition of interest versus subjects without. As a result, the agreement between the same two tests can change (possibly a lot) just by changing the proportion of subjects with and without the condition of interest in the study subject population, even when everything else remains the same. That is, the agreement can change depending on the condition prevalence in the study subject population.
In order to demonstrate this phenomenon, start with the data from Table 6A. The condition prevalence in this study population is 23.2% (51/220). In subjects with the
condition (Reference Standard+ column), the overall percent agreement between the new test and the non-reference standard is 88.2% ((39+6)/51), and in subjects without the condition (Reference Standard− column) it is 98.2% ((1+165)/169. The overall percent agreement combining subjects with and without the condition is 95.9% ((39+6+1+165)/220), which is the same number computed from Table 4. The positive percent agreement is 90.9% (40/(40+4)), and the negative percent agreement is 97.2% (171/(171+5)).
To show how condition prevalence affects agreement, suppose that the condition prevalence in the study population is much lower, but the agreement between the new test and non-reference standard in subjects with and without the condition remains the same. For example, suppose the study population included 676 subjects without the condition (four times 169) instead of 169 subjects so that the condition prevalence in the study population is 7% (51/(51+676)) rather than 23.2%. The new data would look like Table 6C. The Reference standard+ column in Table 6C is the same as Table 6A, but the Reference standard- column in Table 6C is four times the results in Table 6A.
Table 6C. A three-way presentation of results comparing the new test, the non- reference standard, and reference standard. Condition prevalence is one-fourth of that in Table 6A
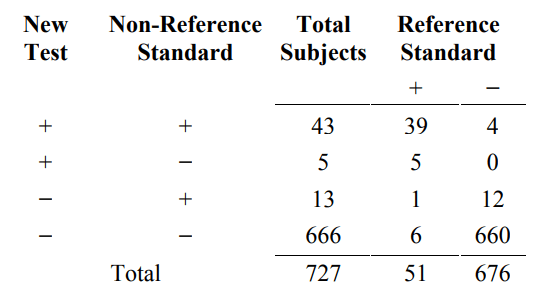
From Table 6C, the percent agreement between the new test and the non-reference standard for subjects with the condition is still 88.2% ((39+6)/51), and for subjects without the condition (Reference Standard− column), it is still 98.2% ((4+660)/676. However, the overall percent agreement combining subjects with and without the condition is 97.5% ((39+6+4+660)/727), higher than the original 95.9%. Showing a more dramatic difference, positive percent agreement is much lower at 76.8% (43/(43+13)) versus 90.9%, and negative percent agreement is slightly higher at 99.2% (666/(666+5)) versus 97.2%.
The performance of the new test and the non-reference standard did not change from Table 6A to 6C, but all of the agreement measures changed simply because the condition prevalence changed. Therefore, it is difficult to generalize agreement measures from Table 4 to another similar subject population unless you have additional information about condition status (such as Table 6A).
7.3 An Example of Discrepant Resolution and its Associated Problems 🔗
As noted before, when a new test is compared to a non-reference standard, the usual calculations from the 2x2 table, a/(a+c) and d/(b+d), respectively, are biased estimates of sensitivity and specificity. Discrepant resolution, described next, is used as an attempt to solve the bias problem. In fact, discrepant resolution does not solve the bias problem; it is just a more complicated wrong solution.
Discrepant resolution is multi-stage testing involving, at a minimum, a new test, a non- reference standard, and a “resolver” test. The decision to use the resolver test depends, in part, on the outcome of the new test. In the discussion below, we assume the resolver is a reference standard since resolving discrepancies using repeat testing by the new test or using a non-reference standard does not provide any useful information about test performance.
In the simplest situation, discrepant resolution can be described as a two-stage testing process in the following manner. Stage 1 involves testing all subjects using the new test and the non-reference standard. The results are presented as in Table 4. In stage 2, when the new test and non-reference standard disagree, an additional test (resolver) is run to see which one is “right.” Table 7 indicates the retested subjects. The outcome of the resolver is reported in Table 8.
Table 7. Two stage testing process of discrepant resolution. The (discrepant) subjects on the off-diagonal (in bold) are additionally tested by a resolver

Table 8. Resolver results
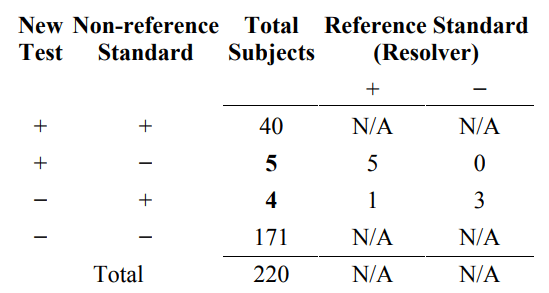
N/A = not available
The results in Table 8 indicate that the new test agrees with the resolver (8 subjects) more than the non-reference standard agrees with the resolver (1 subject) for the study population. However, it is impossible to estimate the relative magnitude of this difference or generalize this difference to a different subject population unless we know the reference standard outcome for all subjects (such as Table 6A) or the condition of interest prevalence in the study population.
From a statistical perspective, retesting discrepant results is not necessary. If you do retest these subjects, FDA recommends reporting these results as in Table 8. However, it is not appropriate to use the resolver results to revise (change) the original 2x2 table of results because the revision is based on assumptions that are not verified and usually are not correct. As a result, it is inappropriate to make sensitivity and specificity-type calculations or agreement calculations using the revised table.
Specifically, it has been the practice of some to revise the original 2x2 table of results (Table 4) based on discrepant resolution (results in Table 8). The original 2x2 table is modified using the following (unsupported) reasoning.
- when the original results (new test and non-reference standard) agree, assume (often incorrectly) that they are both correct and do not make any changes to the table
- when the original results disagree and the non-reference standard disagrees with the resolver, change the non-reference standard result to the resolver result.
Table 9 is an example of how the results from Table 8 are inappropriately used to compute revised results. Specifically, all 40 New Test+/Non-reference Standard+ subjects are incorrectly counted as Reference Standard+, and all 171 New Test−/ Non- reference Standard− subjects are incorrectly counted as Reference Standard−. Next, the 5 New Test+/ Non-reference Standard−/ Reference Standard+ subjects are moved to the New Test+/ Non-reference Standard+ total, and the 3 New Test−/ Non-reference Standard+/ Reference Standard− subjects are moved to the New Test−/ Non-reference Standard− total. The 1 New Test−/ Non-reference Standard+/ Reference Standard+ subject stays in the New Test−/ Non-reference Standard+ total.
Table 9. Inappropriate revision of original results (Table 4) based on discrepant resolution results (Table 8)
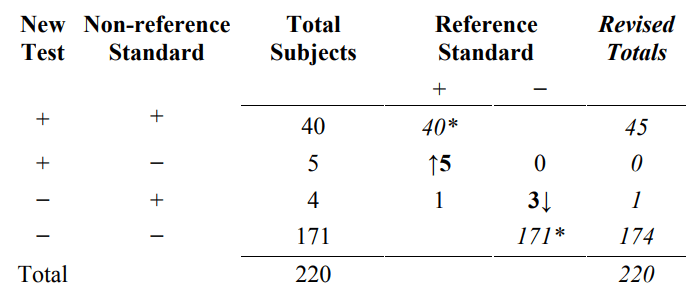
*All subject results incorrectly assumed to be correct (see Table 6A for the correct results for 40* and 171*).
Typically, the revised totals from Table 9 are presented in another 2x2 table such as Table 10B.
Table 10. Inappropriate revised results (Table 10B) based on discrepant resolution of the original results (Table 7)

There are several consequences of revising the original 2x2 table using resolver results. Three consequences are listed below.
- the columns of the revised table are not clearly defined and do not necessarily represent condition status, as assumed
- calculations of sensitivity and specificity from the revised table are not correct
- the “apparent” overall percent agreement calculated from the revised table will always be greater than or equal to percent agreement calculated from the original 2x2 table.
The third consequence needs further explanation. The agreement calculated from the revised results is called “apparent” because agreement with “what” is not clear. For some subjects, it is agreement with the non-reference standard, and for others it is agreement with the reference standard. The reason apparent agreement can only get better is that results can move from the off-diagonal (disagreement) cells to diagonal (agreement) cells in the table, but they cannot move from agreement to disagreement. In fact, using a coin flip as the resolver will also improve apparent agreement. Finally, revising results based on discrepant resolution involves using the outcome of the new unproven test as part of the comparative process used to determine the new test performance. FDA believes this last procedure contradicts good science.
In summary, it is not appropriate to revise the original 2x2 table of results based on discrepant resolution because the revision is based on assumptions that are not verified and usually are not correct. As a result, it is inappropriate to make sensitivity and specificity type calculations or agreement calculations using the revised table. Instead, FDA recommends reporting the original 2x2 table of results (Table 4), a description of the non-reference standard, appropriate agreement measures, and corresponding confidence intervals.
8. Glossary 🔗
biased estimate — estimate that is systematically too high or too low
diagnostic accuracy — the extent of agreement between the outcome of the new test and the reference standard
discrepant resolution — a two-stage testing process that uses a resolver to attempt to classify patients for whom the new test and non-reference standard disagree
external validity — the degree to which results from the study are sufficiently reflective of the “real world” performance of the device in the intended use population
false negative result — a negative test result for a subject in whom the condition of interest is present (as determined by the designated reference standard)
false positive result — a positive test result for a subject in whom the condition of interest is absent (as determined by the designated reference standard)
FN — the number of subjects/specimens with false negative test results
FP — the number of subjects/specimens with false positive test results
intended use population — those subjects/patients (and specimen types) for whom the test is intended to be used; this is called the target population in STARD
likelihood ratio of negative test — the ratio of the true positive rate (sensitivity) and false positive rate (1-specificity); calculated as sensitivity/(1-specificity)
likelihood ratio of positive test — the ratio of the false negative rate (1-sensitivity) and true negative rate (specificity); calculated as (1-sensitivity)/specificity
negative percent agreement (new/non ref. std.) — the proportion of non-reference standard negative subjects in whom the new test is negative
overall agreement — the proportion of subjects in whom the new test and the non- reference standard give the same outcome
positive percent agreement (new/non ref. std.) — the proportion of non-reference standard positive subjects in whom the new test is positive
predictive value of a negative result (sometimes called negative predictive value or NPV) — the proportion of test negative patients who do not have the target condition; calculated as 100xTN/(TN+FN)
predictive value of a positive result (sometimes called positive predictive value or PPV) — the proportion of test positive patients who have the target condition; calculated as 100xTP/(TP+FP)
prevalence — the frequency of a condition of interest at a given point in time expressed as a fraction of the number of individuals in a specified group with the condition of interest compared to the total number of individuals (those with the condition plus those without the condition of interest) in the specified group; pretest probability of the condition of interest in a specified group
reference standard — the best available method for establishing the presence or absence of the target condition; the reference standard can be a single test or method, or a combination of methods and techniques, including clinical follow-up
sensitivity — the proportion of subjects with the target condition in whom the test is positive; calculated as 100xTP/(TP+FN)
specificity — the proportion of subjects without the target condition in whom the test is negative; calculated as 100xTN/(FP+TN)
study population — the subjects/patients (and specimen types) included in the study
target condition (condition of interest) — a particular disease, a disease stage, health status, or any other identifiable condition within a patient, such as staging a disease already known to be present, or a health condition that should prompt clinical action, such as the initiation, modification, or termination of treatment
TN — the number of subjects/specimens with true negative test results
TP — the number of subjects/specimens with true positive test results
true negative result — a negative test result for a subject in whom the condition of interest is absent (as determined by the designated reference standard)
true positive result — a positive test result for a subject in whom the condition of interest is present (as determined by the designated reference standard)
9. References 🔗
Albert, P. S. (2006). Imputation approaches for estimating diagnostic accuracy for multiple tests from partially verified designs. Biometrics (in press). [Available as Technical Report 042, Biometric Research Branch, Division of Cancer Treatment and Diagnosis, National Cancer Institute (http://linus.nci.nih.gov/~brb/TechReport.htm)]
Albert, P.S., & Dodd, L.E. (2004). A cautionary note on the robustness of latent class models for estimating diagnostic error without a gold standard. Biometrics, 60, 427– 435.
Albert, P. S. and Dodd, L. E. (2006). On estimating diagnostic accuracy with multiple raters and partial gold standard evaluation. Journal of the American Statistical Association (in press). [Available as Technical Report 041, Biometric Research Branch, Division of Cancer Treatment and Diagnosis, National Cancer Institute (http://linus.nci.nih.gov/~brb/TechReport.htm)]
Alonzo, T. A., & Pepe, M. S. (1999). Using a combination of reference tests to assess the accuracy of a new diagnostic test. Statistics in Medicine 18, 2987–3003.
Altman, D. G., Machin, D., Bryant, T. N., & Gardner, M. J. (Eds.). (2000). Statistics with confidence, Second Ed. Bristol: British Medical Journal.
Begg, C.G. (1987). Biases in the assessment of diagnostic tests. Statistics in Medicine, 6, 411–423.
Bossuyt, P.M., Reitsma, J.B., Bruns, D.E., Gatsonis, C.A., Glasziou, P.P., Irwig, L.M., Lijmer, J.G., Moher, D., Rennie, D., & deVet, H.C.W. (2003). Towards complete and accurate reporting of studies of diagnostic accuracy: The STARD initiative. Clinical Chemistry, 49(1), 1–6. (Also appears in Annals of Internal Medicine (2003) 138(1), W1–12 and in British Medical Journal (2003) 329(7379), 41–44)
Bossuyt, P.M., Reitsma, J.B., Bruns, D.E., Gatsonis, C.A., Glasziou, P.P., Irwig, L.M., Moher, D., Rennie, D., deVet, H.C.W., & Lijmer, J.G. (2003). The STARD statement for reporting studies of diagnostic accuracy: Explanation and elaboration. Clinical Chemistry, 49(1), 7–18. (Also appears in Annals of Internal Medicine (2003) 138(1), W1–12 and in British Medical Journal (2003) 329(7379), 41–44)
Bross, I. Misclassification in 2x2 tables. Biometrics 1954, 10, 478–86.
CLSI (formerly NCCLS) document EP12-A. (2002). User protocol for evaluation of qualitative test performance: Approved guideline. CLSI, 940 West Valley Road, Suite 1400, Wayne, PA 19087.
CLSI (formerly NCCLS) document GP10-A. Assessment of the clinical accuracy of laboratory tests using receiver operating characteristic (ROC) plots: Approved guideline. CLSI, 940 West Valley Road, Suite 1400, Wayne, PA 19087.
Dunn, G., & Everitt, B. (1995). Clinical biostatistics: An introduction to evidence-based medicine. New York: John Wiley & Sons.
Fleiss, J. L. (1981). Statistical methods for rates and proportions, Second Ed. New York: John Wiley & Sons.
Gart, J.J., & Buck, A.A. (1966). Comparison of a screening test and a reference test in epidemiologic studies. II: A probabilistic model for the comparison of diagnostic tests. American Journal of Epidemiology 83, 593–602.
Green, T.A., Black, C.M., & Johnson, R.E. (1998). Evaluation of bias in diagnostic-test sensitivity and specificity estimates computed by discrepant analysis. Journal of Clinical Microbiology, 36, 375–81.
Hadgu, A. (1996). The discrepancy in discrepant analysis. Lancet, 348, 592–593.
Hagdu, A. (1997). Bias in the evaluation of DNA-amplification tests for detecting
Chlamydia trachomatis. Statistics in Medicine, 16, 1391–9.
Hagdu, A. (1999). Discrepant analysis: A biased and an unscientific method for estimating test sensitivity and specificity. Journal of Clinical Epidemiology, 52, 1231–1237.
Hawkins, D. M., Garrett, J. A., & Stephenson, B. (2001). Some issues in resolution of diagnostic tests using an imperfect gold standard. Statistics in Medicine, 20, 1987– 2001.
Hayden, C. L., & Feldstein, M. L. (2000, Jan/Feb). Dealing with discrepancy analysis part 1: The problem of bias. IVD Technology, 37, 42.
Hayden, C. L., & Feldstein, M. L. (2000, Mar/Apr). Dealing with discrepancy analysis part 2: Alternative analytical strategies. IVD Technology, 51, 57.
Hilden, J. (1997). Discrepant analysis—or behavior? Lancet, 350, 902.
Hilden, J. (1998). Author’s reply: Discrepant analysis and screening for Chlamydia trachomatis. Lancet, 351, 217–218.
Hui, S. L., & Zhou, X. H. (1998). Evaluation of diagnostic tests without gold standards.
Statistical Methods in Medical Research, 7, 354–370.
Kondratovich, Marina (2003). Verification bias in the evaluation of diagnostic devices. Proceedings of the 2003 Joint Statistical Meetings, Biopharmaceutical Section, San Francisco, CA.
Kruskal, W., & Mosteller, F. (1979). Representative sampling, III: The current statistical literature. International Statistical Review, 47, 245–265.
Lang, T. A., & Secic, M. (1997). How to report statistics in medicine. Philadelphia: American College of Physicians.
Lipman, H.B., & Astles, J.R. (1998). Quantifying the bias associated with use of discrepant analysis. Clinical Chemistry, 44, 1, 108–115.
McAdam, A. J. (2000). Discrepant analysis: How can we test a test? Journal of Clinical Microbiology, 38, 2027–2029.
Miller, W.C. (1998a). Bias in discrepant analysis: When two wrongs don’t make a right.
Journal of Clinical Epidemiology, 51, 219–31.
Miller, W.C. (1998b). Editorial response: Can we do better than discrepant analysis for new diagnostic test evaluation? Clinical Infectious Diseases, 27, 1186–93.
Pepe, M. S. (2003). The statistical evaluation of medical tests for classification and prediction. New York: Oxford University Press.
Rothwell, P. M. (2006). Factors that can affect the external validity of randomized controlled trials. PLoS Clinical Trials 1(1): e9.
Saah, A. J., & Hoover, D. R. (1997). “Sensitivity” and “specificity” reconsidered: The meaning of these terms in analytical and diagnostic settings. Annals of Internal Medicine, 126, 91–94.
Schachter, J., Stamm, W.E., & Quinn, T.C. (1998). Editorial response: Discrepant analysis and screening for Chlamydia trachomatis. Lancet, 351, 217–218.
Schatzkin, A., Conner, R.J., Taylor, P.R., & Bunnag, B. (1987). Comparing new and old screening tests when a reference procedure cannot be performed on all screenees.
American Journal of Epidemiology, 125, 672–8.
Shoukri, M.M. (1998). Agreement, Measurement of. Encyclopedia of Biostatisitics.
Armitage, P., & Colton, T. (Eds.). New York: John Wiley & Sons, 103–117.
Staquet, M., Rozencweig, M., Lee, Y.J., & Muggia, F.M. (1981). Methodology for the assessment of new dichotomous diagnostic tests. Journal of Chronic Diseases, 34, 599–610.
Tenenbein, A. (1971). A double sampling scheme for estimating from binomial data with misclassifications: Sample size determination. Biometrics, 27, 935–44.
Thibodeau, L.A. (1981). Evaluating diagnostic tests. Biometrics, 37, 801–804.
Torrance-Rynard, V.L., & Walter, S.D. (1997). Effects of dependent errors in the assessment of diagnostic test performance. Statistics in Medicine, 16, 2157–2175.
Vacek, P.M. (1985). The effect of conditional dependence on the evaluation of diagnostic tests. Biometrics 41, 959–968.
Valenstein, P. (1990). Evaluating diagnostic tests with imperfect standards. American Journal of Clinical Pathology, 93, 252–258.
Walter, S.D., & Irwig, L.M. (1988). Estimation of test error rates, disease prevalence and relative risk from misclassified data: A review. Clinical Epidemiology, 41, 923–37.
Zhou, X. H. (1998). Correcting for verification bias in studies of diagnostic tests.
Statistical Methods in Medical Research, 7, 337–53.
Zhou, X. H., Obuchowski, N. A., & McClish, D. K. (2002). Statistical methods in diagnostic medicine. New York: John Wiley & Sons.
Zweig, M.H., & Campbell, G. (1993). Receiver-operating characteristic (ROC) plots: A fundamental evaluation tool in clinical medicine. Clinical Chemistry, 39, 561–577.
Footnotes 🔗
-
This database is publicly available at www.clsi.org. ↩
-
See www.consort-statement.org/stardstatement.htm or two articles by Bossuyt et al. 2003a,b. ↩
-
You can contact statisticians at the FDA Center for Devices and Radiological Health, Office of Surveillance and Biometrics, Division of Biostatistics, at (240) 276-3133. ↩
-
By “ambiguous” we mean any results that are intermediate, inconclusive, incomplete, uninterpretable, unsatisfactory, unavailable, in a “gray zone,” or otherwise anything different than either positive or negative. ↩
-
By “equivocal” we mean any results that your test, when used as per the instructions, classifies as equivocal, indeterminate, intermediate, gray-zone, or anything else different than either positive or negative. ↩
