
Innolitics introduction 🔗
Innolitics provides US FDA regulatory consulting to startups and established medical-device companies. We’re experts with medical-device software, cybersecurity, and AI/ML. See our services and solutions pages for more details.
We have practicing software engineers on our team, so unlike many regulatory firms, we speak both “software” and “regulatory”. We can guide your team through the process of writing software validation and cybersecurity documentation and we can even accelerate the process and write much of the documentation for you (see our Fast 510(k) Solution).
About this Transcript 🔗
This document is a transcript of an official FDA (or IMDRF) guidance document. We transcribe the official PDFs into HTML so that we can share links to particular sections of the guidance when communicating internally and with our clients. We do our best to be accurate and have a thorough review process, but occasionally mistakes slip through. If you notice a typo, please email a screenshot of it to Miljana Antic at mantic@innolitics.com so we can fix it.
Preamble 🔗
This document supersedes the Special 510(k) content in “The New 510(k) Paradigm - Alternate Approaches to Demonstrating Substantial Equivalence in Premarket Notifications,” issued on March 20, 1998.
Document issued on September 13, 2019.
The draft of this document was issued on September 28, 2018.
For questions about this document regarding CDRH-regulated devices, contact ORP: Office of Regulatory Programs/ DRP1: Division of Submission Support/ Premarket Notification and Classification Team at 510K_Program@fda.hhs.gov or 301-796-5640. For questions regarding this document regarding CBER-regulated devices, contact the Office of Communication, Outreach and Development (OCOD) in CBER at 1-800-835-4709 or 240-402-8010 or by email at ocod@fda.hhs.gov.
Contains non-binding guidance.
I. Introduction 🔗
This guidance provides the Food and Drug Administration’s (FDA) current thinking on premarket notifications (510(k)s) appropriate for review as a Special 510(k). The intent of this guidance is to describe an optional pathway for certain well-defined device modifications where a manufacturer modifies its own legally marketed device, and design control procedures produce reliable results that can form, in addition to other 510(k) content requirements, the basis for substantial equivalence (SE). This guidance clarifies the types of technological changes appropriate for review as Special 510(k)s. Specifically, within the scope of appropriate changes, we are including certain design and labeling changes, including changes to the indications for use, by focusing on whether the method(s) to evaluate the change(s) are well-established, and whether the results can be sufficiently reviewed in a summary or risk analysis format.
The Special 510(k) Program is consistent with FDA’s statutory mission to protect and promote human health and FDA’s commitment to helping patients gain timely access to new medical devices that are high quality, safe and effective by using efficient review practices consistent with least burdensome principles.1 This guidance, provides consistency, clarity, and transparency to industry to describe when a Special 510(k) is appropriate. This guidance supersedes the Special 510(k) policy in the “The New 510(k) Paradigm: Alternate Approaches to Demonstrating Substantial Equivalence in Premarket Notifications.”
For the current edition of the FDA-recognized standard(s) referenced in this document, see the FDA Recognized Consensus Standards Database Web site at https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfStandards/search.cfm. For more
information regarding use of consensus standards in regulatory submissions, please refer to FDA guidance titled Appropriate Use of Voluntary Consensus Standards in Premarket Submissions for Medical Devices2 and Standards Development and the Use of Standards in Regulatory Submission Reviewed in CBER.3
FDA's guidance documents, including this guidance, do not establish legally enforceable responsibilities. Instead, guidances describe the Agency’s current thinking on a topic and should be viewed only as recommendations, unless specific regulatory or statutory requirements are cited. The use of the word should in Agency guidance means that something is suggested or recommended, but not required.
II. Background 🔗
FDA established the Special 510(k) Program in 1998 and described the program and policy in the guidance document “The New 510(k) Paradigm: Alternate Approaches to Demonstrating Substantial Equivalence in Premarket Notifications” (“New 510(k) Paradigm Guidance”).4 The program was intended to create an efficient review process for certain changes subject to 510(k) submission requirements.
Design controls were added to the Quality System (QS) Regulation and have been in effect since June 1, 1997 (21 CFR 820.30, 61 FR 52602). The Special 510(k) Program leverages design controls requirements to support SE determinations through the reliance on risk analysis and verification and validation for existing devices. Special 510(k)s allow FDA and industry to rely on previous Agency review of detailed information, where appropriate, without altering any statutory or regulatory requirements related to the premarket notification process under sections 510 and 513 of the FD&C Act, and 21 CFR 807 Subpart E. The Special 510(k) Program provides a least burdensome approach to the review of certain changes to a manufacturer’s own legally marketed predicate device (“existing device”) because a Special 510(k) provides an efficient pathway for manufacturers to provide the minimum required information necessary to establish SE for a modified device. Because of this efficiency, FDA stated in the New 510(k) Paradigm Guidance that we intend to process Special 510(k)s within 30 days of receipt by the Document Control Center, rather than the 90 days for 510(k)s required by section 510(n)(1) of the FD&C Act.
The Special 510(k) Program was previously limited to review of changes that did not affect the device’s intended use nor alter the device’s fundamental scientific technology. Under this approach, Special 510(k)s that included modifications to the indications for use or any labeling change that affected the device’s intended use and/or modifications that had the potential to alter the fundamental scientific technology of the device compared to the manufacturer’s own legally marketed predicate device5 were routinely converted to Traditional 510(k)s. FDA now no longer intends to focus on changes that affect indications for use or alter fundamental scientific technology in determining whether the 510(k) is appropriate as a Special 510(k). Instead, FDA’s approach focuses on whether the method(s) to evaluate the change(s) are well-established, and whether the results can be sufficiently reviewed in a summary or risk analysis format. A Special 510(k) would generally not be appropriate for devices that manufacture a biological product at the point of care, because there would likely be no well-established method to evaluate such changes and/or the performance data would not be reviewable in a summary or risk-analysis format.
Through the finalization of this guidance, we are updating the Special 510(k) Program to clarify existing policy and the types of changes appropriate for the program to improve the efficiency of 510(k) review. Under this approach, certain changes to the indications for use may be made. FDA has also clarified the types of changes to technological characteristics that are appropriate for review as a Special 510(k). For more information about how FDA evaluates whether changes to the indications for use fall within the same intended use and how differences in technology affect FDA’s SE determination process, see the FDA guidance document The 510(k) Program: Evaluating Substantial Equivalence in Premarket Notifications [510(k)].6 Special 510(k)s remain subject to the content and format requirements for 510(k) submissions, 510(k) summary or 510(k) statement, and class III certifications (21 CFR 807.87, 807.90, 807.92, 807.93, and 807.94, respectively).
III. Special 510(k) Program 🔗
The Special 510(k) Program is intended to facilitate the submission, review, and clearance of a change to a manufacturer’s own legally marketed predicate device (“existing device”) that is already authorized for commercial distribution through 510(k) clearance, preamendments status, reclassification, or through a granted De Novo classification request under section 513(f)(2) of the FD&C Act.
For certain device changes, FDA believes that design control procedures can produce reliable results that can form the basis for a SE determination without compromising the statutory and regulatory criteria for SE. Under design controls, manufacturers are required to conduct verification and validation (21 CFR 820.30(f) and (g)). Verification and validation include procedures to ensure that design outputs meet design inputs, and that devices conform to defined user needs and intended uses. The QS Regulation, 21 CFR Part 820, has records establishment and maintenance requirements that apply to design changes subject to design controls (21 CFR 820.30 and 820.180). These records must be made available to an FDA investigator upon request under section 704(e) of the FD&C Act.
When a manufacturer considers submitting a Special 510(k), FDA recommends that manufacturers consider all relevant guidance documents, special controls, or recognized voluntary consensus standards that apply to the device type or to a scientific topic area (e.g., biocompatibility or electromagnetic compatibility). For example, if a manufacturer is modifying a powered lower extremity exoskeleton device, then the manufacturer’s design inputs should address the special controls that FDA has established for that device type under 21 CFR 890.3480. If a manufacturer modifies an in vitro diagnostic (IVD), the manufacturer’s design inputs should include any relevant clinical and laboratory standards recognized by FDA. This guidance is not intended to supersede device-specific policies regarding the submission of complete test reports or Special 510(k) considerations that are identified in some guidance documents. For example, as discussed in the FDA guidance Reprocessing Medical Devices in Health Care Settings: Validation Methods and Labeling (referred to as Reprocessing Guidance),7 510(k) submissions for certain reusable devices are required to include validation data pursuant to section 510(q) of the FD&C Act. These devices are identified in FDA’s Federal Register notice published in 82 FR 268078 and Appendix E of the Reprocessing Guidance. FDA does not consider such 510(k) submissions to be appropriate for review under the Special 510(k) Program because these validation data reports cannot be provided in a summary or risk analysis format.
Subject to FDA’s acceptance review in accordance with the guidance Refuse to Accept Policy for 510(k)s,9 FDA generally reviews Special 510(k) submissions within 30 days of receipt. If a manufacturer submits a Special 510(k) that FDA does not believe is appropriate for review under the Special 510(k) Program, FDA intends to convert the submission to a Traditional 510(k) and notify the submitter.
When FDA converts a Special 510(k) to a Traditional 510(k), management concurrence occurs prior to the conversion. During FDA’s notification of 510(k) conversion, FDA intends to provide an explanation of the reason(s) for conversion using the Special 510(k) factors discussed below. The 510(k)-conversion process can result in delayed review because complete test reports are not reviewed in a Special 510(k), but are typically requested in a Traditional 510(k). This difference in content between Special and Traditional 510(k)s often results in FDA refusing to accept the 510(k) after conversion to a Traditional 510(k). Therefore, FDA recommends that both FDA and manufacturers consider the below factors to determine whether review as a Special 510(k) is appropriate. If the 510(k) submission was accepted for a substantive review and later converted to a Traditional 510(k), the review clock continues into FDA’s 90-day statutory deadline under section 510(n)(1) of the FD&C Act and remains subject to MDUFA performance goals for 510(k) submissions.
In accordance with 21 CFR 807.81(a)(3), and as explained in FDA’s guidance Deciding When to Submit a 510(k) for a Change to an Existing Device10 (“510(k) Modifications Guidance”), not all changes require a new 510(k) and manufacturers should use a risk-based assessment approach, as appropriate, to guide their analysis of whether a new 510(k) is likely required. If a manufacturer determines that a new 510(k) is likely required, then the flowchart provided in Figure 1 and the companion text guide FDA staff and manufacturers through the decision-making process to determine whether a particular submission is appropriate for review as a Special 510(k).
Subject to the framework identified in sections III.A-E of this guidance, a design or labeling change to an existing device (including certain changes to the indications for use) may be appropriate for a Special 510(k) when:
- The proposed change is submitted by the manufacturer legally authorized to market the existing device;
- Performance data are unnecessary, or if performance data are necessary, well-established methods are available to evaluate the change; and
- All performance data necessary to support SE can be reviewed in a summary or risk analysis format.
These considerations and associated decision making are summarized in Figure 1. Examples of changes that are and are not appropriate for review under the Special 510(k) Program are included in Appendix B.
Although most Class I devices are not subject to the design control requirements of the QS Regulation, manufacturers of Class I (reserved) devices11 may voluntarily elect to comply with the design controls regulation and submit Special 510(k)s.
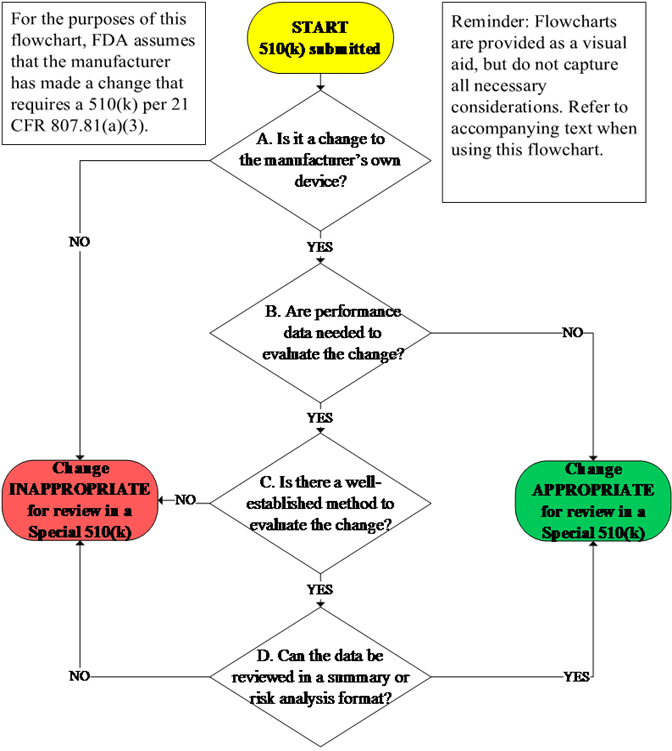
A. Is it a change to the manufacturer’s own device? 🔗
To be within the scope of the Special 510(k) Program, the 510(k) should be for a change to the submitter’s own legally marketed predicate device. This is because the Special 510(k) Program relies on the Agency’s previous review of detailed information and a manufacturer who modifies its own legally marketed device is able to conduct the risk analysis and the necessary verification and validation activities to demonstrate that the design outputs of the modified device meet the design input requirements in a Special 510(k) submission. FDA intends to convert Special 510(k)s to Traditional 510(k)s when the submitter is not the manufacturer legally authorized to market the predicate device. In cases where the referenced 510(k) was submitted under a different name than the submitter, FDA recommends that the submitter include a statement affirming that they are the manufacturer legally authorized to market the predicate device.
B. Are performance data needed to evaluate the change? 🔗
Manufacturers should use their design control procedures and consider the information necessary to support SE to determine whether performance data are needed to evaluate the change. As part of design controls, manufacturers must establish and maintain procedures for the validation, or where appropriate, verification, of design changes before their implementation (21 CFR 820.30(i)). Verification and validation testing, however, may not be necessary to support SE. For example, FDA may receive a 510(k) from a manufacturer requesting clearance to label their device as Magnetic Resonance (MR) Unsafe after previously labeling their device as ‘Safety in MR Imaging Not Evaluated.’ As discussed in the FDA guidance document Establishing Safety and Compatibility of Passive Implants in the Magnetic Resonance (MR) Environment,12 MR Unsafe labeling is based on a scientific rationale and does not involve any performance data. In other cases, verification and validation testing may be necessary to support changes in indications for use and design. For example, identification of a new environment of use in the indications for use or labeling without changes to the intended users or user interface may result in the need for additional verification and validation testing to support continued electromagnetic compatibility (EMC) and other performance characteristics.
In cases where manufacturers determine under their design control procedures that no additional verification or validation testing is necessary to evaluate a change that otherwise requires submission and clearance of a 510(k), manufacturers may submit these changes as a Special 510(k) with a clear rationale supporting their conclusion that no performance data are necessary. When FDA does not agree with the manufacturer’s assessment about whether performance data will be necessary to support a SE determination, FDA intends to continue its review with the additional Special 510(k) factors discussed in sections III.C-E before considering whether the 510(k) submission should be converted to a Traditional 510(k).
C. Is there a well-established method to evaluate the change? 🔗
FDA believes that in order to qualify for the Special 510(k) Program, well-established methods should be available to evaluate the change under design controls. The Special 510(k) Program should not include the submission and review of complete test reports, but summary information generated from well-established methods. Well-established methods are those that have been established for evaluation of the device, device type, or scientific topic area, and are validated according to scientific principles. Minor deviations to a well-established method may be acceptable within the context of a Special 510(k), but significant deviations to the protocol or acceptance criteria of a well-established method can result in the 510(k) being no longer appropriate for review as a Special 510(k). If manufacturers are uncertain whether protocol or acceptance criteria deviations from an otherwise well-established method are significant, they can use the Pre-Submission process to obtain feedback per the FDA guidance Requests for Feedback and Meetings for Medical Device Submissions: The Q-Submission Program.13
FDA believes that well-established methods may include:
- The submitter’s methods, protocols, and acceptance criteria used to support the previously cleared 510(k) that can be applied to the subject 510(k);
- Methods found in an FDA-recognized voluntary consensus standard14 or FDA guidance document;
- Qualified medical device development tools (MDDTs); or
- Widely available and accepted methods published in the public domain, scientific literature, or found acceptable by FDA through the submitter’s own 510(k)-clearance, a granted De Novo classification request, or premarket application (PMA) approval.
FDA recommends that manufacturers describe why the methods applied to evaluate the impact of the changes included in a Special 510(k) are well-established. This description can include a discussion that the methods and acceptance criteria were the same as the predicate device and are relevant to the change under review. When standards undergo revision, the FDA-recognized version(s) as identified in our online database15 are considered to include well-established methods. Such methods should rely on established acceptance criteria, or a comparison of performance to the predicate device and/or reference device16 under the same testing methodology. For example, Traditional 510(k)s often identify the verification and validation approaches that are used for software such that many subsequent software changes may occur under a Special 510(k). To remain appropriate for review as a Special 510(k), all test methods used to support the 510(k) should be well-established.
Submissions that use methods that rely on clinical studies or animal data to support SE are not typically appropriate for the Special 510(k) Program because the methodologies and endpoints vary, are often dependent on the condition(s) being studied, and cannot be appropriately summarized. The use of clinical specimens to conduct IVD verification and validation does not necessarily mean that a well-established method does not exist to evaluate the change. When FDA does not agree that a well-established method exists to evaluate the change, FDA intends to convert the Special 510(k) to a Traditional 510(k). In that case, FDA intends to explain to the submitter why the method to evaluate the change is not well-established.
D. Can the data be reviewed in a summary or risk analysis format? 🔗
To be appropriate for a Special 510(k), the results from verification and validation associated with design or labeling changes should be able to be placed in a summary or risk analysis format without losing information necessary to support SE. Complete test reports should not be submitted in a Special 510(k). If complete test reports are submitted, FDA intends to assess whether the information can be reviewed in a summary format before converting to a Traditional 510(k). This assessment should occur during FDA’s acceptance review in accordance with the 510(k) Refuse to Accept (RTA) policy. Given the shorter timeframe for review of Special 510(k)s, if the submitter cannot provide summary information within the timeframe identified during interactive RTA review, FDA intends to convert the submission to a Traditional 510(k).
FDA does not believe that data can be summarized when the SE determination will depend on the Agency’s interpretation of the underlying data, such as images, raw graphs, or line item data. For example, FDA does not believe that data can be placed in a summary format when fatigue to failure testing involves the review of graphical images to interpret the failure modes observed. In limited circumstances where a small number of representative images for non-clinical performance are submitted, such would be appropriate for a Special 510(k). For example, representative images used to demonstrate radiopacity for guidewires or devices with radiopaque markers may be included in a Special 510(k). FDA has included anticipated common scenarios for when data may be unable to be summarized without loss of information in section III.E.
FDA believes that the results from risk management activities, including relevant verification and validation information, produced under design controls procedures can be used to support a SE determination of the Special 510(k) under the conditions described in this guidance. As described in Appendix A, this information should include a concise summary of design control activities and verification and validation testing required to comply with 21 CFR 820.30 based on a manufacturer’s procedures. To have sufficient information to establish SE under a Special 510(k), your summary or table should describe, for each change that required a 510(k), the specific verification and validation activities, how the methods applied are appropriate for the change, acceptance criteria, any changes or deviations from testing methods in previous 510(k) submissions, and a summary of the results. When FDA does not agree that the performance data can be summarized, FDA intends to convert the submission to a Traditional 510(k). This should typically occur during the RTA review. In this case, FDA intends to explain to the submitter why the performance data could not be provided in a summary or risk analysis format.
In accordance with the flexibility of the QS Regulation, there can be different approaches to the summary of design control activities and verification and validation that can be included in a Special 510(k). This can include redlined software requirements specification (SRS) and design documentation that clearly documents the changes that were made, consistent with well-established methods. Manufacturers can include their risk management documentation, such as a Design Failure Modes and Effects Analysis (DFMEA), along with a separate summary of supporting verification and validation. Manufacturers could also summarize their risk management activities with the specifics of verification and validation that provide information necessary for FDA’s SE determination process. To facilitate FDA review, the summary of design control activities and verification and validation should highlight and focus on the information that is relevant to the changes under review. FDA has provided examples in Appendix C of this guidance.
E. Additional considerations 🔗
Because FDA intends to review a Special 510(k) within 30 days, FDA believes there are some circumstances when it is not appropriate to submit a Special 510(k), including:
- When evaluation of the change(s) to the device generally involve greater than three scientific disciplines (e.g., biocompatibility, sterility, electromagnetic compatibility);
- For multiple devices with unrelated changes as described in the FDA guidance Bundling Multiple Devices or Multiple Indications in a Single Submission;17
- When a recent QS inspection has resulted in the issuance of a violative inspection report identifying observations related to design controls that are relevant to the design changes under review in the 510(k). If a manufacturer believes such violations are unrelated to the subject 510(k), they should provide a rationale for why the 510(k) should still be appropriate for review under the Special 510(k) Program;
- When Special 510(k)s are submitted for common scenarios that FDA anticipates a review of complete test reports will be necessary to establish SE, such as:
- Changes to the indications for use that are supported by clinical, animal,18 or cadaver data;
- Use of novel sterilization methods as described in the FDA guidance Submission and Review of Sterility Information in Premarket Notification (510(k)) Submissions for Devices Labeled as Sterile;19
- Changes to introduce initial MR Conditional labeling, or significant deviations from the test methods used to establish MR Conditional labeling in the original 510(k);
- Change from single-use to reusable when reprocessing validation or human factors data should be provided; and
- Use of analytical chemistry testing using International Organization for Standardization (ISO) 10993-1820 and/or toxicological risk assessment using ISO 10993-1721 to address biocompatibility.22
- For a reprocessed single-use device (SUD) that requires the submission of cleaning, sterilization, and functional performance validation data under section 510(o) of the FD&C Act and in FDA’s Federal Register notice published in 70 FR 5691123 requiring the submission of SUD validation data. Consistent with the FDA guidance Medical Device User Fee and Modernization Act of 2002, Validation Data in Premarket Notification Submissions (510(k)s) for Reprocessed Single-Use Medical Devices,24 if the reprocessed SUD does not require validation data, and is otherwise appropriate for a Special 510(k) submission, the reprocessor may submit a Special 510(k); and
- For changes that could affect the reprocessing of reusable devices required by section 510(q) of the FD&C Act to include reprocessing validation in 510(k) These devices are identified in FDA’s Federal Register notice published in 82 FR 2680725 and Appendix E of the Reprocessing Guidance.26
Appendix A. Recommended content of a Special 510(k) 🔗
A Special 510(k) should include:
- A coversheet clearly identifying the submission as a “Special 510(k): Device Modification;”
- The name of the manufacturer’s legally marketed (existing) device and the 510(k) number under which it was cleared;
- Information required under 21 CFR 807.87, including a description of the modified device, a comparison to the cleared device, the indications for use of the device, and the proposed labeling for the To help ensure that FDA has a complete understanding of the device under review, this should include:
- A detailed description of the change(s) made to the device that resulted in the submission of a new 510(k). When labeling or specific technological characteristics (e.g., materials, dimensions) are unchanged in comparison to the predicate, the submission should clearly state that no changes were made;
- A comparison of the modified device to the cleared device in a tabular format;
- Clean and redlined copies of documents that were updated from what was submitted in the predicate device’s submission because of the device change (e.g., labeling, risk analysis); and
- Other changes to labeling or design since the most recently cleared 510(k) (i.e., those that did not require submission of a new 510(k)) that would have been documented as part of the original 510(k), in accordance with the recommendations in the FDA guidance Deciding When to Submit a 510(k) for a Change to an Existing Device.27
- If the Special 510(k) includes reference(s) or a declaration of conformity to a recognized voluntary consensus standard, we recommend that you consult the FDA guidance Appropriate Use of Voluntary Consensus Standards in Premarket Submissions for Medical Devices;28
- A concise summary of the design control activities. Appendix C provides examples of narratives and a table of this information that has been historically provided. FDA considers the information generated from the design control activities to be “appropriate supporting data” within the meaning of 21 CFR 807.87(g). Your risk management file may already contain some of the design control activities in a risk analysis format. In lieu of creating a new table that addresses all recommended content, you may instead submit your risk analysis as an attachment or appendix to your This summary should include the following:
- Identification of the risk analysis method(s) used to assess the impact of the change on the device and the results of the analysis;
- Identification of the device change(s);
- Identification of all risks associated with each device change, including identification of risks that are considered new because of the change; and
- Risk control measures to mitigate identified risks (e.g., labeling, verification).
- Based on the risk analysis, an identification of the verification and/or validation activities required to comply with 21 CFR 820.30. This identification should include a summary of test methods, acceptance criteria, and results, and why each is adequate to establish If unchanged from a previous premarket submission, the manufacturer can reference the location of protocols and acceptance criteria by providing a submission and section numbers. When the results are quantitative in nature, the submission should include basic descriptive statistics, such as the mean, standard deviation, and range of the data. Protocol deviations observed during testing should be provided and justified, if applicable. When appropriate, the summary of verification and validation should include:
- For non-standardized test methods only:
- A reference to the protocol used for the existing device with an identification of any differences (e.g., protocol, test conditions, pre- defined acceptance criteria, sample size) from the previous 510(k). If protocol changes were made, the results summary should describe why the test methods, acceptance criteria, and results support SE.
- For test methods described in an FDA-recognized standard:
- Cross-reference to the relevant section of the Special 510(k) where a declaration of conformity (DOC) was submitted under section 514(c) of the FD&C Act. This should also include or cross-reference applicable supplemental documentation per ISO/IEC 17050-229 to support the DOC; or
- If a DOC is not submitted, the basis for general use of a consensus standard should include underlying information or data that supports how the standard was For Special 510(k)s, submitters that rely on general use of a consensus standard should provide a description of methods with deviations, selected options and the reasons for their selection, acceptance criteria, and a results summary. See the FDA guidance Appropriate Use of Voluntary Consensus Standards in Premarket Submissions for Medical Devices30 for more information about the use of voluntary consensus standards.
- For non-standardized test methods only:
- Indications for Use form (Form FDA 3881);31 and
- A signed statement by the manufacturer’s designated individual(s) responsible for design control activities that includes:
- A statement that, as required by the risk analysis, all design verification and validation activities were performed by the designated individual(s) and the results demonstrated that the predetermined acceptance criteria were met; and
- A statement that the submitter has complied and is not currently in violation of the design control procedure requirements as specified in 21 CFR 820.30 and the records are available for review, upon request.
Appendix B. Examples of changes 🔗
These examples are for illustrative purposes and may not include all details for each change. The examples are intended to help FDA staff and industry determine which changes can be submitted as a Special 510(k).
Example B.1
Change: The submitter wants to change their 2-D chest x-ray image processing software to add a feature that highlights nodules in the lung. The submitter is also requesting to modify their indications for use to describe this new software feature that now quantifies and characterizes information about the nodules.
Relevant Questions:
A - Is it a change to the manufacturer’s own device?
Yes, the submitter is the manufacturer of the predicate device.
B - Are performance data needed to evaluate the change?
Yes. Clinical testing should be provided to support marketing clearance for such a change in the indications for use to assess the performance of the software on patients with and without nodules in the lung. This clinical testing should support that the software can successfully quantify and characterize information about the nodules.
C - Is there a well-established method to evaluate the change?
No. There are no well-established methods identified in the predicate’s submission for the evaluation of lung nodules, consensus standards, or widely available and accepted methods published in the public domain to address the change in the indications for use.
D - Can the data be reviewed in a summary or risk analysis format?
N/A.
Decision: Change cannot be reviewed in a Special 510(k).
Example B.2
Change: The submitter wants to add wireless control capabilities to their bilevel positive airway pressure (BiPAP) device intended to treat patients with obstructive sleep apnea.
Relevant Questions:
A - Is it a change to the manufacturer’s own device?
Yes, the submitter is the manufacturer of the predicate device.
B - Are performance data needed to evaluate the change?
Yes. The predicate device did not contain and was not tested for wireless functionality. Verification and validation should be conducted to ensure that the BiPAP has acceptable wireless quality of service, coexistence, cybersecurity, and maintains EMC in its intended environment of use, as described in the FDA guidance Radio Frequency Wireless Technology in Medical Devices.32
C - Is there a well-established method to evaluate the change?
No. International Electrotechnical Commission (IEC) 60601-1-233 and Association for the Advancement of Medical Instrumentation (AAMI) Technical Information Report (TIR) 6934 can be used to support EMC and wireless coexistence. However, there are not well- established methods in an FDA-recognized voluntary consensus standard or in the manufacturer’s previous 510(k) that address the methods to evaluate the addition of wireless control for this BiPAP. The test methods vary depending on the wireless quality of service necessary for the device’s intended use and environment of use.
D - Can the data be reviewed in a summary or risk analysis format?
N/A.
Decision: Change cannot be reviewed in a Special 510(k).
Example B.3
Change: The submitter wants to modify their general indications for delivering illumination and laser energy for photocoagulation to include specific clinical applications for treatment of retinopathy.
Relevant Questions:
A - Is it a change to the manufacturer’s own device?
Yes, the submitter is the manufacturer of the predicate device.
B - Are performance data needed to evaluate the change?
Yes. Clinical testing is typically provided to support marketing clearance for such a change in the indications for use. The requested change in the indications for use now identify a specific disease condition. The clinical outputs have changed from general coagulation of blood vessels to treatment of retinopathy. Clinical testing should be conducted to assess new outcomes such as decrease in vision impairment, whereas the predicate assessed the general outcome of successful vessel coagulation.
C - Is there a well-established method to evaluate the change?
No. There is no well-established method identified in the predicate’s submission or a consensus standard to evaluate clinical endpoints for this device. The SE determination rests on a review of the underlying clinical performance data.
D - Can the data be reviewed in a summary or risk analysis format?
N/A.
Decision: Change cannot be reviewed in a Special 510(k).
Example B.4
Change: The submitter currently markets a cardiac output monitor that is cleared for use with their endotracheal tube. The submitter is requesting clearance to modify the indications for use so that the submitter’s cardiac output monitor can be used with their 510(k)-cleared endobronchial tube that also includes integrated electrodes for sensing.
Relevant Questions:
A - Is it a change to the manufacturer’s own device?
Yes, the submitter is the manufacturer of the predicate device.
B - Are performance data needed to evaluate the change?
Yes. Verification should be completed to demonstrate that the newly identified tube can be used for cardiac output by impedance cardiography as safely and effectively with the monitor as the endotracheal tube does with the monitor, and that the monitor and endobronchial tube both continue to function as intended.
C - Is there a well-established method to evaluate the change?
Yes. Because the bench testing to verify the change uses the same protocol as the predicate device, and the methods and acceptance criteria have not changed, the protocol is considered a well-established method. In addition, this type of connection for the specified tube and monitor has been included in other cleared 510(k) submissions for this device, and the submitter referenced these devices in their submission.
D - Can the data be reviewed in a summary or risk analysis format?
Yes. The protocol, methods and acceptance criteria were not modified from those used in the predicate submission to evaluate the change. The existing methods were appropriate to evaluate the change because the same cardiac output parameters are intended to be monitored and displayed. The acceptance criteria and a summary of the results were provided for each test. The results can be summarized because the SE determination does not depend on the Agency’s interpretation of the underlying data, such as images, raw graphs, or line item data.
Decision: Change can be reviewed in a Special 510(k).
Example B.5
Change: The company is requesting clearance to change the environment of use identified in their labeling for their transcutaneous electrical nerve stimulation (TENS) device from a professional healthcare facility only to both professional healthcare facility and home use. The device is still intended to be used under the direction and supervision of a healthcare professional.
Relevant Questions:
A - Is it a change to the manufacturer’s own device?
Yes, the submitter is the manufacturer of the predicate device.
B - Are performance data needed to evaluate the change?
Yes. There are different acceptance criteria for electrical safety and electromagnetic compatibility (EMC) to address home use.
C - Is there a well-established method to evaluate the change?
Yes. For example, the FDA-recognized standard methods American National Standards Institute (ANSI)/AAMI ES60601-135 and IEC 60601-2-1036 address basic safety and essential performance, EMC (IEC 60601-1-237), and basic safety for home use devices (ANSI/AAMI HA60601-1-1138 or IEC 60601-1-1139), along with the International Special Committee on Radio Interference (CISPR) 1140 emission limits for Group 1 and Class B. The manufacturer provided their statement of essential performance and associated device-specific acceptance criteria.
D - Can the data be reviewed in a summary or risk analysis format?
Yes. The particular standard used was identified. The acceptance criteria and results were summarized in a tabular format. A justification was provided for all results that were outside the bounds of an acceptance range or differed from the predicate. The results can be summarized because the SE determination does not depend on the Agency’s interpretation of the underlying data, such as images, raw graphs, or line item data.
Decision: Change can be reviewed in a Special 510(k).
Example B.6
Change: The submitter is requesting clearance to market metal bone screws terminally sterilized via gamma irradiation that were previously only supplied non-sterile and sterilized by the end user. The indications for use and materials of construction remain unchanged from the clearance for the manufacturer’s existing device.
Relevant Questions:
A - Is it a change to the manufacturer’s own device?
Yes, the submitter is the manufacturer of the predicate device.
B - Are performance data needed to evaluate the change?
Yes. The submitter should include an evaluation of biocompatibility, sterility, pyrogenicity, package integrity, and shelf-life to support the proposed change. Non-clinical testing to address performance of the device outside of biocompatibility, sterility, packaging, and shelf-life is not necessary based on a scientifically-based rationale from the submitter that gamma irradiation does not impact the material composition or properties of this metallic device.
Based on the recommendations in the FDA guidance Use of International Standard ISO 10993-1, “Biological evaluation of medical devices - Part 1: Evaluation and testing within a risk management process,”41 the submitter provided a valid scientifically-based rationale supporting the decision that no further biocompatibility testing was necessary to address this change.
C - Is there a well-established method to evaluate the change?
Yes. The FDA guidance Submission and Review of Sterility Information in Premarket Notification (510(k)) Submissions for Devices Labeled as Sterile42 indicates that gamma irradiation is an Established Sterilization Method, Established Category A. The FDA- recognized standards ISO 11137-143 and ISO 11137-244 can be used to support the sterilization validation. Pyrogenicity can be assessed using the recommendations discussed in the FDA guidance documents Submission and Review of Sterility Information in Premarket Notification (510(k)) Submissions for Devices Labeled as Sterile45 and Pyrogen and Endotoxins Testing: Questions and Answers,46 and the methods described in the FDA- recognized versions of ANSI/AAMI ST7247 and United States Pharmacopeia (USP) <161>.48 Package integrity and shelf-life for this change can be evaluated through accelerated aging using American Society for Testing and Materials (ASTM) F198049 and package integrity testing for visual integrity, seal integrity, and seal strength using the methods identified in ASTM F1886/F1886M,50 ASTM F2096,51 and ASTM F88/F88M,52 respectively.
D - Can the data be reviewed in a summary or risk analysis format?
Yes. The methods are standardized, and the results can be summarized because the SE determination does not depend on the Agency’s interpretation of the underlying data, such as images, raw graphs, or line item data. The FDA guidance Submission and Review of Sterility Information in Premarket Notification (510(k)) Submissions for Devices Labeled as Sterile53 discusses how sterilization validation, package integrity, and pyrogenicity information can be summarized in 510(k) submissions.
Decision: Change can be reviewed in a Special 510(k).
Example B.7
Change: The submitter wants to increase the number of channels for their receive-only magnetic resonance (MR) coil.
Relevant Questions:
A - Is it a change to the manufacturer’s own device?
Yes, the submitter is the manufacturer of the predicate device.
B - Are performance data needed to evaluate the change?
Yes. Consistent with the FDA guidance Submission of Premarket Notifications
for Magnetic Resonance Diagnostic Devices,54 performance testing should be provided for the increased number of coils to address image quality metrics and patient safety from surface heating. For a receive-only coil, this should include signal-to-noise ratio, image uniformity, and coil surface heating assessments.
C - Is there a well-established method to evaluate the change?
Yes. There are standard test methods for MR devices such as FDA-recognized consensus standards National Electrical Manufacturers Association (NEMA) MS 955 and NEMA MS
6.56 The predicate device used the same standards, protocols, and acceptance criteria.
D - Can the data be reviewed in a summary or risk analysis format?
Yes. The methods can be summarized and the results can be placed into a summary format for each test conducted because the SE determination does not depend on the Agency’s interpretation of the underlying data, such as images, raw graphs, or line item data. While a small, representative subset of sample images were included, the manufacturer did not include a complete dataset of images that would be necessary for FDA to evaluate SE. Instead, the manufacturer provided a statement from a U.S. Board Certified radiologist attesting that images produced by the device are of sufficient quality for diagnostic use.
Decision: Change can be reviewed in a Special 510(k).
Example B.8
Change: The submitter wants to add analytical sensitivity data for the new H7N9 influenza strain to their diagnostic test.
Relevant Questions:
A - Is it a change to the manufacturer’s own device?
Yes, the submitter is the manufacturer of the predicate device.
B - Are performance data needed to evaluate the change?
Yes. Analytical reactivity testing should be provided to address the addition of analytical sensitivity data for the new strain into the labeling.
C - Is there a well-established method to evaluate the change?
Yes. The same protocol as the original submission was used for collecting and assessing the data. The acceptance criteria were not altered from those used for the original device. No additional types of evaluation are needed.
D - Can the data be reviewed in a summary or risk analysis format?
Yes. The results can be summarized because the SE determination does not depend on the Agency’s interpretation of the underlying data, such as images, raw graphs, or line item data. In addition, the methods and acceptance criteria are unmodified from the predicate testing.
Decision: Change can be reviewed in a Special 510(k).
Example B.9
Change: The submitter wants to change the labeling of their blade-form endosseous dental implant from “Safety in MRI Not Evaluated” to “MR Conditional.”
Relevant Questions:
A - Is it a change to the manufacturer’s own device?
Yes, the submitter is the manufacturer of the predicate device.
B - Are performance data needed to evaluate the change?
Yes. Non-clinical performance testing to support SE should be provided by manufacturers seeking MR Conditional labeling for a device that contains metallic components. The FDA guidance document Establishing Safety and Compatibility of Passive Implants in the Magnetic Resonance (MR) Environment57 provides recommendations for such testing.
C - Is there a well-established method to evaluate the change?
Yes. There are FDA-recognized voluntary consensus standards such as ASTM F2503,58 ASTM F2052,59 ASTM F2213,60 ASTM F2182,61 and ASTM F211962 for MR compatibility
testing of passive implants.
D - Can the data be reviewed in a summary or risk analysis format?
No. Although there are consensus standards for all test methods, FDA does not believe this data can be summarized because the SE determination will depend on FDA’s interpretation of the underlying data to support the MR Conditional label. This includes interpretation of device-specific pass/fail criteria and results that are not addressed in the standard. This is referenced in section III.E as an anticipated common scenario for when data may be unable to be summarized.
Decision: Change cannot be reviewed in a Special 510(k).
Example B.10
Change: The submitter wants to increase the size of their MR Conditional blade-form endosseous dental implant from 4mm long to 5mm long.
Relevant Questions:
A - Is it a change to the manufacturer’s own device?
Yes, the submitter is the manufacturer of the predicate device.
B - Are performance data needed to evaluate the change?
Yes. FDA has designated special controls for blade-form endosseous dental implants in 21 CFR 872.3640(b)(2)(i)-(ix) that must be addressed, including performance testing for fatigue, corrosion, biocompatibility evaluation, sterility, and evaluation of the device in the MR environment. The FDA guidance document Establishing Safety and Compatibility of Passive Implants in the Magnetic Resonance (MR) Environment63 recommends that manufacturers seeking MR Conditional labeling for a device that contains metallic components provide
non-clinical performance testing to support SE. The manufacturer also submitted a biocompatibility evaluation based on a scientific justification.
C - Is there a well-established method to evaluate the change?
There are FDA-recognized voluntary consensus standards such as ASTM F2503,64 ASTM F2052,65 ASTM F2213,66 ASTM F2182,67 and ASTM F211968 for MR compatibility testing of passive implants. There are also FDA-recognized voluntary consensus standards for fatigue testing of endosseous dental implants, such as ANSI/American Dental Association (ANSI/ADA) Standard No. 12769 and ISO 1480170 to address the performance of the device. In addition, ISO 14801 and ANSI/ADA Standard No. 127 are applicable to all dental implants.
D - Can the data be reviewed in a summary or risk analysis format?
Yes. There are consensus standards for test methods, and guidance documents for reference. The fatigue testing can be placed into a summary format because the size change does not necessitate protocol or acceptance criteria deviations. In addition, the size change (4mm to 5mm) does not necessitate clinical or animal data. Because there has been no material change, and the 1 mm size change is not expected to alter the safety of the device with respect to MR compatibility, and the protocol and acceptance criteria has not changed, the MR testing results can be placed into a summary format because the SE determination does not depend on the Agency’s interpretation of the underlying data, such as images, raw graphs, or line item data.
Decision: Change can be reviewed in a Special 510(k).
Example B.11
Change: The submitter proposes to change the shape of the test cassette for a lateral flow immunoassay for fecal occult blood. The new test cassette has a longer and slimmer housing design in comparison to the predicate device.
Relevant Questions:
A - Is it a change to the manufacturer’s own device?
Yes, the submitter is the manufacturer of the predicate device.
B - Are performance data needed to evaluate the change?
Yes. A method comparison study should be conducted using the predicate device and candidate device to measure patient samples from the intended use population.
C - Is there a well-established method to evaluate the change?
Yes. The submitter stated in their submission that the test method used is the same as that used for assessment of the predicate device.
D - Can the data be reviewed in a summary or risk analysis format?
Yes. Given that the test method used is the same for this submission as used for the predicate, a risk analysis was used to assess the impact of the change on the device and its components. The results of the method comparison study can be reviewed in terms of meeting the predefined acceptance criteria and a summary of study results that includes the observed false positive and false negative rates.
Decision: Change can be reviewed in a Special 510(k).
Example B.12
Change: The submitter wants to change their over-the-counter (OTC) human chorionic gonadotropin (hCG) urine pregnancy test device to add an absorbent sample application tip and change the instructions for use to specify that the results should be read between three and ten minutes after use, instead of at five minutes.
Relevant Questions:
A - Is it a change to the manufacturer’s own device?
Yes, the submitter is the manufacturer of the predicate device.
B - Are performance data needed to evaluate the change?
Yes. Analytical validation studies and a clinical lay user study should be provided to demonstrate that device performance is substantially equivalent to the predicate.
C - Is there a well-established method to evaluate the change?
Yes. The protocols used for the analytical validation and clinical studies (method comparison study and lay user study) were consistent with protocols that have been found acceptable by FDA in the submitter’s own 510(k) submission for the predicate device.
D - Can the data be reviewed in a summary or risk analysis format?
Yes. For the analytical and clinical validation studies, the Agency’s determination of substantial equivalence relies on well-defined acceptance criteria that are specific enough to assess whether the device has substantially equivalent performance to its predicate. For the analytical method comparison study, results can be reviewed as a summary of false positive and false negative rates. For the clinical lay user study, the patient population information can be adequately described in the study protocol and the results can be reviewed as a summary of agreement rate between the subject and predicate devices. These assessments do not require review of study line item data.
Decision: Change can be reviewed in a Special 510(k).
Example B.13
Change: The submitter wants to widen the hematocrit range for blood samples that their OTC blood glucose meter can measure and change the design and materials used in the external housing of their device.
Relevant Questions:
A - Is it a change to the manufacturer’s own device?
Yes, the submitter is the manufacturer of the predicate device.
B - Are performance data needed to evaluate the change?
Yes. Analytical validation should be conducted to assess the effect of the wider hematocrit range on device performance. Disinfection efficacy studies and a cleaning and disinfection robustness study should assess whether the new external case design and materials can be adequately cleaned and disinfected.
C - Is there a well-established method to evaluate the change?
Yes. The FDA guidance documents Self-Monitoring Blood Glucose Test Systems for Over- the-Counter Use71 and Blood Glucose Monitoring Test Systems for Prescription Point-of- Care Use72 include recommendations for hematocrit range evaluation, disinfection efficacy, and cleaning and disinfection robustness testing methods.
D - Can the data be reviewed in a summary or risk analysis format?
Yes. For these analytical validation studies, the Agency’s determination of substantial equivalence relies on well-defined acceptance criteria that are specific enough to assess whether the device has substantially equivalent performance to its predicate. For the hematocrit and cleaning and disinfection robustness studies, summary results of the measurement bias can be reviewed. For the disinfection efficacy study, results can be reviewed as a pass/fail summary compared to predefined acceptance criteria for viral inactivation. These assessments do not require review of study line data.
Decision: Change can be reviewed in a Special 510(k).
Example B.14
Change: The submitter wants to change the Reference Range section in the package insert for an immunoglobulin light chain specific assay by adding the normal range of the kappa lambda free light chain ratio. The submitter is not proposing to change their indications for use or device design.
Relevant Questions:
A - Is it a change to the manufacturer’s own device?
Yes. The submitter is the manufacturer of the predicate device.
B - Are performance data needed to evaluate the change?
Yes. Testing with relevant samples should be completed to determine the normal range of the test results.
C - Is there a well-established method to evaluate the change?
Yes. The reference range study may be conducted based on the FDA-recognized version of Clinical and Laboratory Standards Institute (CLSI) EP28-A3c.73
D - Can the data be reviewed in a summary or risk analysis format?
Yes. A summary of subject demographic information and summary statistics (e.g., mean, median, and range) can be reviewed for the samples used in the new reference range study.
Decision: Change can be reviewed in a Special 510(k).
Example B.15
Change: The submitter wants to change the threshold for the deoxyribonucleic acid (DNA) probe of a specific bacterial pathogen to address customer feedback. The threshold change increases the stringency for determining if a test result is positive to reduce potential false positives and as a preventative measure to mitigate against variability at customers’ sites. No changes were made to the assay reagents or procedure.
Relevant Questions:
A - Is it a change to the manufacturer’s own device?
Yes. The submitter is the manufacturer of the predicate device.
B - Are performance data needed to evaluate the change?
Yes. Analytical testing, including both reanalysis of existing data and new performance “wet” testing, should be conducted to demonstrate that the assay performance is not negatively impacted by the change.
C - Is there a well-established method to evaluate the change?
Yes. The analytical testing methods and acceptance criteria are the same as the predicate device’s submission.
D - Can the data be reviewed in a summary or risk analysis format?
Yes. Summary review of limited analytical performance testing are sufficient to determine the assay performance is not adversely affected by the change.
Decision: Change can be reviewed in a Special 510(k).
Appendix C. Examples of the summary of design control activities 🔗
This section provides sample design control activities summaries that can be used to support a Special 510(k). Because of the inherent flexibility of design controls and the QS regulation, this summary may differ depending on a manufacturer’s internal procedures. The examples are intended to show different formats that have been used in previously cleared Special 510(k) submissions.
Example C.1
In the subject 510(k), the manufacturer requested clearance to change their lacrimal stent to remove a metal ring, change the shape of the stent’s duct tube, and alter the surface area of a hydrophilic coating. The manufacturer’s design controls narrative described that a risk analysis was conducted to assess the impact of the changes on the subject device using internal design control procedures and a fault tree analysis described in the FDA-recognized version of ISO 14971.74 The manufacturer included their fault tree analysis specific to this design change in an attachment for the Special 510(k) to identify the hazardous situations, causes, risk control measures, and acceptability before and after risk control measures. The manufacturer explained that the protocol, test methods, and acceptance criteria used were the same as those used in the predicate submission and provided references to the applicable sections in the previous submission. The risk analysis identified the verification and validation activities necessary to establish SE, and summarized that information in the following table:
Table 1. Example design control activities summary for a hypothetical lacrimal stent
| Device Change | Risks | Verification/Validation Method(s) | Acceptance Criteria | Summary of Results |
|---|---|---|---|---|
|
Removal of ring |
Damaged tissue Damage to device during insertion with bougie causes delay in patient treatment |
Penetration test performed with bougie (Protocol and acceptance criteria same as Kxxxxxx without any deviations) |
Breaking load shall be greater than 9N | Pass (12/12) Mean: 15.0 Standard deviation: 0.39 Range: 14.4-15.6 |
|
Shape change |
Damaged tissue Damage to device causes delay in patient treatment |
Simulated insertion test with bougie Bending test with bougie (Protocol and acceptance criteria same as Kxxxxxx without any deviations) |
For both tests, visual inspection shall demonstrate that the device can be inserted without damage. | Pass (20/20) for both tests |
|
Change in hydrophilic coating surface area |
Difficulty inserting causes delay in patient treatment Abnormalities on catheter causes damage to tissue |
Insertion test with simulated lacrimal duct Visual inspection (Protocol and acceptance criteria same as Kxxxxxx without any deviations) |
No visual damage after simulated insertion No droplets, extraneous matter, or abnormalities are visualized under a microscope |
Pass (15/15) Pass (10/10) |
| Adverse tissue reaction from material coating area and geometric changes. | Biocompatibility evaluation in agreement with recommendations in Use of International Standard ISO 10993-1, “Biological evaluation of medical devices - Part 1: Evaluation and testing within a risk management process” (CDRH’s Biocompatibility Guidance).1 Leveraged all biocompatibility testing from another device with similar type and duration of contact, greater surface area, and same formulation and processing by the same device manufacturer. |
Materials of construction and manufacturing materials do not introduce chemicals that raise a biocompatibility concern. | Biocompatibility testing is not needed because device does not introduce a biocompatibility risk. | |
| Materials of construction and manufacturing materials do not introduce chemicals that raise a material- mediated pyrogenicity concern. | Material- mediated pyrogenicity testing is not needed because device does not introduce a material- mediated pyrogenicity risk. |
Example C.2
In the subject 510(k), the manufacturer requested clearance to modify the geometric design and constructive materials of the single-use sheath used in a self-retaining retractor for neurosurgery. The manufacturer’s design controls narrative described that a design failure modes and effects analysis (DFMEA) was included in the submission. In accordance with their risk management procedures, the manufacturer identified their design inputs, identified risks with their evaluation, risk control measures, and residual risk. The risk analysis identified the verification and validation activities and summarized them in this table:
Table 2. Example design control activities summary for a hypothetical sheath
| Device Change | Risks | Verification/Validation Method(s) | Acceptance Criteria | Summary of Results | |
|---|---|---|---|---|---|
|
Material change to polyethylene |
Adverse tissue reaction from material change |
Biocompatibility evaluation in agreement with recommendations in CDRH’s Biocompatibility Guidance.1 Based on our risk management procedures, biocompatibility testing was repeated for some endpoints. |
Cytotoxicity (ISO 10993-5)2 using the ISO minimum essential medium (MEM) Elution method. The protocol used the same test article preparation and extraction conditions as the predicate (MEM with 10% serum, 37 ºC, 24 hours, at a surface area/volume ratio of 6 cm2/ml), appropriate controls, extracts were not stored for more than 24 hours, and were not altered (e.g., filtered or pH adjusted). These testing conditions are the same as the predicate device, the extracts did not change color, appear turbid or have particulates, and there were no deviations/amendments from the protocol. |
Reactivity grade shall be 0, which is the same as for the predicate device. | There was no evidence of the test extract causing cell lysis or toxicity (Grade = 0) for three replicates at 48 hours. Latex Positive Control = Grade 3 High Density Polyethylene Negative Control = Grade 0 The test article is non-cytotoxic. |
| Irritation (ISO 10993-10)3 using the intracutaneous reactivity method. The protocol used the same test article preparation and extraction conditions as the predicate (saline and sesame seed oil extract solvents, 50 ºC, 72 hours, at a surface area/volume ratio of 6 cm2/ml), appropriate controls, extracts were not stored for more than 24 hours, and extracts were not altered (e.g., filtered or pH adjusted). These testing conditions are the same as the predicate device, the extracts did not change color, appear turbid, or have particulates, and there were no deviations/amendments from the protocol. |
The difference between the mean reaction score for the test article and control shall be ≤1.0, which is the same as the predicate device. | The polar extract showed no irritation (Grade 0) and the non- polar extract showed minimal irritation (Grade 0/1) at 24, 48 and 72 hours, which was consistent with the negative vehicle control results. Saline Vehicle Control = Grade 0 at all timepoints Sesame Vehicle Control = Grade 0/1 at all timepoints No adverse in vivo findings were noted in any of the test or control animals. The test article is a non-irritant. |
|||
|
Sensitization (ISO 10993- 10)4 using the guinea pig maximization test. The protocol used the same test article preparation and extraction conditions as the predicate (saline and sesame oil extract solvents, 50 ºC, 72 hours, at a surface area/volume ratio of 6 cm2/ml), appropriate controls, extracts were not stored for more than 24 hours, and extracts were not altered (e.g., filtered or pH adjusted). These testing conditions are the same as the predicate device, the extracts did not change color, appear turbid or have particulates, and there were no deviations/amendments from the protocol. |
Grade 0 in both test and control animals, which is the same as the predicate device. | Both the polar and non-polar extracts scored 0 at 24 and 48 hours for all test subjects, which was consistent with the negative control. The extracts did not change color or have particulates. The test article is a non-sensitizer. |
|||
|
Acute systemic toxicity Reviewed: 1) Literature; and 2) SDS that are in accordance with Appendix D of 29 CFR 1910.1200.5 |
Materials of construction and manufacturing materials do not introduce chemicals that elicit acute systemic toxicity. SDS meets 29 CFR 1910.1200 content. |
Acute systemic toxicity testing is not needed because device does not introduce an acute systemic toxicity risk. Neither analytical chemistry testing nor a toxicological risk assessment were used to support this rationale. | |||
|
Material-mediated pyrogenicity Leveraged material-mediated pyrogenicity testing from another polyethylene device with similar type and duration of contact, greater surface area, and same formulation and processing by the same device owner. |
Materials of construction and manufacturing materials do not introduce chemicals that raise a material- mediated pyrogenicity concern. | Material-mediated pyrogenicity testing is not needed because device does not introduce a material-mediated pyrogenicity risk. Neither analytical chemistry testing nor a toxicological risk assessment were used to support this rationale. | |||
| Patient infection Device failure causes patient injury or delay in procedure. |
Sterilization validation was completed using an established method (gamma irradiation) in conformity with ISO 11137-1 without deviation.6 The sterilization validation approach was Verification Dose Maximum (VDmax) for a Sterility Assurance Level (SAL) of 10-6 in accordance with AAMI TIR33.7 Package integrity testing was also conducted using methods consistent with the predicate device (seal integrity, dye penetration, and visual inspection). |
Devices shall maintain package integrity and have SAL of 10-6. |
Package integrity testing results all passed (n=30 each). Bioburden studies passed. Sterilization validation established SAL of 10-6. |
||
| Geometric design change | Damage to devices causes patient injury or delay in procedure. | Specification review and dimensional analysis. | Dimensional verification shall demonstrate that the sheath geometric change does not interfere with obturator. |
Pass (n=20) | |
| Adverse tissue reaction from geometric shape change. | |||||
| Design validation to confirm that the sheath continues to meet manufacturer-defined user requirements. Simulated-use testing was conducted with a prospective user to confirm that the device can achieve its intended use. (Protocol and acceptance criteria same as Kxxxxxx without any deviations) |
The sheath shall be able to be used with third- party accessories and provide access to the tissue identified in labeling. | Pass | |||
|
Implantation and thrombogenicity Reviewed geometric changes per CDRH’s Biocompatibility Guidance8 (Attachment A, Table A.1) to determine whether implantation or thrombogenicity (which can be impacted by geometry) are recommended for this device type/duration of contact. |
For externally communicating devices in contact with tissue or bone for < 24 hours, Table A.1 indicates that implantation and thrombogenicity assessments are not necessary. |
Additional biocompatibility evaluation to assess the impact of the geometric change on the biological response is not needed. |
“Use of International Standard ISO 10993-1, ‘Biological evaluation of medical devices - Part 1: Evaluation and testing within a risk management process,’” available at: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/use-international-standard-iso-10993-1-biological-evaluation-medical- devices-part-1-evaluation-and.↩︎
ISO 10993-5 Biological evaluation of medical devices - Part 5: Tests for in vitro cytotoxicity.↩︎
ISO 10993-10 Biological evaluation of medical devices - Part 10: Tests for irritation and skin sensitization.↩︎
ISO 10993-10 Biological evaluation of medical devices - Part 10: Tests for irritation and skin sensitization.↩︎
For more information about Safety Data Sheets, see 77 FR 17574.↩︎
ISO 11137-1 Sterilization of health care products – Radiation – Part 1: Requirements for the development, validation and routine control of a sterilization process for medical devices.↩︎
AAMI TIR33 Sterilization of health care products — Radiation — Substantiation of a selected sterilization dose — Method VDmax.↩︎
“Use of International Standard ISO 10993-1, ‘Biological evaluation of medical devices - Part 1: Evaluation and testing within a risk management process,’” available at: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/use-international-standard-iso-10993-1-biological-evaluation-medical- devices-part-1-evaluation-and.↩︎
Footnotes 🔗
-
Section 1003 of the Federal Food, Drug, and Cosmetic Act (FD&C Act). ↩
-
https://www.fda.gov/regulatory-information/search-fda-guidance-documents/appropriate-use-voluntary-consensus- standards-premarket-submissions-medical-devices. ↩
-
https://www.fda.gov/regulatory-information/search-fda-guidance-documents/standards-development-and-use- standards-regulatory-submissions-reviewed-center-biologics-evaluation. ↩
-
The New 510(k) Paradigm Guidance was superseded by this guidance and “The Abbreviated 510(k) Program.” ↩
-
A legally marketed predicate device is a device that was legally marketed prior to May 28, 1976 (i.e., preamendments), reclassified from class III to class II or class I, found substantially equivalent through a 510(k), or granted marketing authorization through the De Novo classification process. ↩
-
https://www.fda.gov/regulatory-information/search-fda-guidance-documents/510k-program-evaluating-substantial- equivalence-premarket-notifications-510k. ↩
-
https://www.fda.gov/regulatory-information/search-fda-guidance-documents/reprocessing-medical-devices-healthcare-settings-validation-methods-and-labeling. ↩
-
June 9, 2017. ↩
-
https://www.fda.gov/regulatory-information/search-fda-guidance-documents/refuse-accept-policy-510ks. ↩
-
https://www.fda.gov/regulatory-information/search-fda-guidance-documents/deciding-when-submit-510k-changeexisting-device. ↩
-
See section 510(l) of the FD&C Act. ↩
-
https://www.fda.gov/regulatory-information/search-fda-guidance-documents/establishing-safety-and- compatibility-passive-implants-magnetic-resonance-mr-environment. ↩
-
https://www.fda.gov/regulatory-information/search-fda-guidance-documents/requests-feedback-and-meetings- medical-device-submissions-q-submission-program. ↩
-
For the purposes of this guidance, FDA-recognized voluntary consensus standards include those that FDA has recognized or decided to recognize. For more information, see “Appropriate Use of Voluntary Consensus Standards in Premarket Submissions for Medical Devices,” available at https://www.fda.gov/regulatory-information/search- fda-guidance-documents/appropriate-use-voluntary-consensus-standards-premarket-submissions-medical-devices. ↩
-
https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfStandards/search.cfm. ↩
-
Consistent with “The 510(k) Program: Evaluating Substantial Equivalence in Premarket Notifications [510(k)]” (https://www.fda.gov/regulatory-information/search-fda-guidance-documents/510k-program-evaluating-substantial- equivalence-premarket-notifications-510k), reference devices are other legally marketed devices that may be used to support scientific methodology or standard reference values for Decisions 5a and 5b of the 510(k) decision-making flowchart after a manufacturer successfully navigates through Decision Point 4 using a single predicate device. ↩
-
https://www.fda.gov/regulatory-information/search-fda-guidance-documents/bundling-multiple-devices-or- multiple-indications-single-submission. ↩
-
FDA supports the principles of the “3R’s,” to reduce, refine, and replace animal use in testing when feasible. We encourage sponsors to consult with us if it they wish to use a non-animal testing method they believe is suitable, adequate, validated, and feasible. We will consider if such an alternative method could be assessed for equivalency to an animal test method. ↩
-
https://www.fda.gov/regulatory-information/search-fda-guidance-documents/submission-and-review-sterility- information-premarket-notification-510k-submissions-devices-labeled. ↩
-
ISO 10993-18 Biological evaluation of medical devices – Part 18: Chemical characterization of materials. ↩
-
ISO 10993-17 Biological evaluation of medical devices – Part 17: Establishment of allowable limits for leachable substances. ↩
-
We recognize that chemical information may be used to support another part of your biocompatibility evaluation (e.g., a rationale for why a specific biocompatibility test is not needed). Use of chemical information (e.g., literature, Safety Data Sheet (SDS)) that does not involve a toxicological risk assessment may be acceptable. For more information, see Example C.2. ↩
-
This notice was published on September 29, 2005. ↩
-
https://www.fda.gov/regulatory-information/search-fda-guidance-documents/medical-device-user-fee-and- modernization-act-2002-validation-data-premarket-notification. ↩
-
This notice was published on June 9, 2017. ↩
-
https://www.fda.gov/regulatory-information/search-fda-guidance-documents/reprocessing-medical-devices- health-care-settings-validation-methods-and-labeling. ↩
-
https://www.fda.gov/regulatory-information/search-fda-guidance-documents/deciding-when-submit-510k-change- existing-device. ↩
-
hhttps://www.fda.gov/regulatory-information/search-fda-guidance-documents/appropriate-use-voluntary- consensus-standards-premarket-submissions-medical-devices. ↩
-
ISO 17050-2 Conformity assessment – Part 2: Supporting documentation for the general requirements and supporting documentation. ↩
-
https://www.fda.gov/regulatory-information/search-fda-guidance-documents/appropriate-use-voluntary- consensus-standards-premarket-submissions-medical-devices. ↩
-
https://www.fda.gov/regulatory-information/search-fda-guidance-documents/radio-frequency-wirelesstechnology-medical-devices-guidance-industry-and-fda-staff. ↩
-
IEC 60601-1-2 Medical electrical equipment - Part 1-2: General requirements for basic safety and essential performance - Collateral Standard: Electromagnetic disturbances - Requirements and tests. ↩
-
AAMI TIR69 Risk management of radio-frequency wireless coexistence for medical devices and systems. ↩
-
ANSI/AAMI ES60601-1 Medical electrical equipment - Part 1: General requirements for basic safety and essential performance. ↩
-
IEC 60601-2-10 Medical electrical equipment - Part 2-10: Particular requirements for the basic safety and essential performance of nerve and muscle stimulators. ↩
-
IEC 60601-1-2 Medical electrical equipment - Part 1-2: General requirements for basic safety and essential performance - Collateral Standard: Electromagnetic disturbances - Requirements and tests. ↩
-
ANSI/AAMI HA60601-1-11 Medical electrical equipment Part 1-11: General requirements for basic safety and essential performance - Collateral Standard: Requirements for medical electrical equipment and medical electrical systems used in the home healthcare environment. ↩
-
IEC 60601-1-11 Medical electrical equipment - Part 1-11: General requirements for basic safety and essential performance - Collateral Standard: Requirements for medical electrical equipment and medical electrical systems used in the home healthcare environment. ↩
-
CISPR 11 Industrial, scientific and medical equipment – Radio-frequency disturbance characteristics – Limits and methods of measurement. ↩
-
https://www.fda.gov/regulatory-information/search-fda-guidance-documents/use-international-standard-iso- 10993-1-biological-evaluation-medical-devices-part-1-evaluation-and. ↩
-
https://www.fda.gov/regulatory-information/search-fda-guidance-documents/submission-and-review-sterility- information-premarket-notification-510k-submissions-devices-labeled. ↩
-
ISO 11137-1 Sterilization of health care products - Radiation - Part 1: Requirements for development, validation and routine control of a sterilization process for medical devices. ↩
-
ISO 11137-2 Sterilization of health care products - Radiation - Part 2: Establishing the sterilization dose. ↩
-
https://www.fda.gov/regulatory-information/search-fda-guidance-documents/submission-and-review-sterility- information-premarket-notification-510k-submissions-devices-labeled. ↩
-
https://www.fda.gov/regulatory-information/search-fda-guidance-documents/pyrogen-and-endotoxins-testing- questions-and-answers. ↩
-
ANSI/AAMI ST72 Bacterial endotoxins - Test methods, routine monitoring, and alternatives to batch testing. ↩
-
USP <161> Medical Devices - Bacterial Endotoxin and Pyrogen Tests. ↩
-
ASTM F1980 Standard guide for accelerated aging of sterile barrier systems for medical devices. ↩
-
ASTM F1886/F1886M Standard test method for determining integrity of seals for flexible packaging by visual inspection. ↩
-
ASTM F2096 Standard test method for detecting gross leaks in packaging by internal pressurization (bubble test). ↩
-
ASTM F88/F88M Standard test method for seal strength of flexible barrier materials. ↩
-
https://www.fda.gov/regulatory-information/search-fda-guidance-documents/submission-and-review-sterility- information-premarket-notification-510k-submissions-devices-labeled. ↩
-
https://www.fda.gov/regulatory-information/search-fda-guidance-documents/submission-premarket-notifications- magnetic-resonance-diagnostic-devices. ↩
-
NEMA MS 9 Standards Publication Characterization of Phased Array Coils for Diagnostic Magnetic Resonance Images. ↩
-
NEMA MS 6 Determination of Signal-to-Noise Ratio and Image Uniformity for Single-Channel Non-Volume Coils in Diagnostic MR Imaging. ↩
-
https://www.fda.gov/regulatory-information/search-fda-guidance-documents/establishing-safety-and- compatibility-passive-implants-magnetic-resonance-mr-environment. ↩
-
ASTM F2503 Standard Practice for Marking Medical Devices and Other Items for Safety in the Magnetic Resonance Environment. ↩
-
ASTM F2052 Standard Test Method for Measurement of Magnetically Induced Displacement Force on Medical Devices in the Magnetic Resonance Environment. ↩
-
ASTM F2213 Standard Test Method for Measurement of Magnetically Induced Torque on Medical Devices in the Magnetic Resonance Environment. ↩
-
ASTM F2182 Standard Test Method for Measurement of Radio Frequency Induced Heating On or Near Passive Implants During Magnetic Resonance Imaging. ↩
-
ASTM F2119 Standard Test Method for Evaluation of MR Image Artifacts from Passive Implants. ↩
-
https://www.fda.gov/regulatory-information/search-fda-guidance-documents/establishing-safety-and- compatibility-passive-implants-magnetic-resonance-mr-environment. ↩
-
ASTM F2503 Standard Practice for Marking Medical Devices and Other Items for Safety in the Magnetic Resonance Environment. ↩
-
ASTM F2052 Standard Test Method for Measurement of Magnetically Induced Displacement Force on Medical Devices in the Magnetic Resonance Environment. ↩
-
ASTM F2213 Standard Test Method for Measurement of Magnetically Induced Torque on Medical Devices in the Magnetic Resonance Environment. ↩
-
ASTM F2182 Standard Test Method for Measurement of Radio Frequency Induced Heating On or Near Passive Implants During Magnetic Resonance Imaging. ↩
-
ASTM F2119 Standard Test Method for Evaluation of MR Image Artifacts from Passive Implants. ↩
-
ANSI/ADA Standard No. 127 Fatigue Testing for Endosseous Dental Implants. ↩
-
ISO 14801 Dentistry - Implants - Dynamic fatigue test for endosseous dental implants. ↩
-
https://www.fda.gov/regulatory-information/search-fda-guidance-documents/self-monitoring-blood-glucose-test- systems-over-counter-use-0. ↩
-
https://www.fda.gov/regulatory-information/search-fda-guidance-documents/blood-glucose-monitoring-test- systems-prescription-point-care-use. ↩
-
CLSI EP28-A3c Defining, Establishing, and Verifying Reference Intervals in the Clinical Laboratory. ↩
-
ANSI/AAMI/ISO 14971 Medical devices - Application of risk management to medical devices. ↩
