What Happened to Kintsugi 🔗
Kintsugi Health just shut down and open-sourced their voice biomarker AI — a $30 million gift to the global mental health community. Four years of FDA engagement. $16 million spent on regulatory work alone. They never even got to file their De Novo.
Respect for Kintsugi:
They did the right thing by trying to find a legitimate pathway. They did the right thing by open-sourcing their algorithms on Hugging Face so society could still benefit. They were in it to do the right thing — to follow the law, to pay their dues to the US public. Grace Chang went to the Digital Health Advisory Committee and argued their case directly: the process is too burdensome. That takes courage.
Sadness for Kintsugi:
They ran out of funding before they could cross the finish line. Had they just played the non-device CDS exemption game, marketed as a "general wellness" tool, or slapped a "not intended for diagnosis or treatment" disclaimer — like so many others — their trajectory might have been completely different. Three cop-outs, all legal (some more than others), all widely used, and all allowing companies to put effectively clinical tools in front of patients without clinical evidence. Kintsugi didn't take any of them. FDA wants to see companies succeed, I believe that. But the pre-market cost is too steep. This only adds to the fear and uncertainty that is driving an industry-wide stalemate right now. And now, a team of bright innovators will think twice before ever building another regulated medical device. They'll tell their friends. Those bright minds will go to fintech, or enterprise SaaS, or anywhere else. The talent drain is undeniable.
And here's something that doesn't get talked about enough: the benefit-risk analysis shouldn't only consider the risks of the device itself. It should also consider the risk of the device's absence — not just to the individual who would have used it, but to the public at large. Every month a mental health screening tool doesn't reach the market, there are patients who go undetected. Every year a digital therapeutic is stuck in regulatory limbo, there are people who don't get treatment. The cost of inaction isn't zero. It's invisible, but it's enormous. When the system — and by system I mean the entire complex of industry, regulators, and payers, not any one party — kills a device that could have helped millions, that's a public health consequence too — and it should weigh on the other side of the scale.
History Rhymes 🔗

Woebot Health — $123.5 million raised, FDA Breakthrough Device Designation granted, Stanford-trained clinical team, 1.5 million users. Shut down their consumer app June 2025. Founder Alison Darcy told STAT the cost and challenge of FDA authorization made it unsustainable. (Note: Woebot Health as a corporate entity may still exist in some form, but its core product — the app that 1.5 million people used — is gone.)
Pear Therapeutics — the prescription digital therapeutics pioneer. Three FDA-authorized products (reSET, reSET-O, Somryst). Went public via SPAC at a $1.6 billion valuation. Reported $12.7 million in revenue against $136 million in expenses. Filed Chapter 11 in April 2023. Assets sold at auction for $6.05 million.
Akili Interactive — FDA De Novo authorization for EndeavorRx, the first-ever prescription video game for ADHD. Raised $230 million in venture funding plus $163 million from their Nasdaq listing. By Q2 2023, they reported $114,000 in quarterly revenue against $15.3 million in expenses. Laid off 46% of their workforce. Acquired for $0.43 per share and delisted in 2024.
Better Therapeutics — FDA authorized their diabetes management app (AspyreRx) via De Novo in July 2023. Even received a Breakthrough Device Designation for liver disease in February 2024. By March 2024 — eight months after authorization — they terminated all employees and delisted from Nasdaq. They had reported just $4.2 million in cash at year-end 2023, and it wasn't enough.
Every single one tried to do it right.
And here's the part that should concern everyone: even the ones that made it through FDA still died.
Pear Therapeutics had three FDA-authorized products. Akili Interactive had an authorized product. Better Therapeutics had an authorized product. These weren't just companies — they could’ve been the first to make an impact. The first prescription video game for ADHD. The first FDA-authorized digital therapeutic for substance use disorder. The first prescription digital therapeutic for type 2 diabetes. These authorizations were historic. We may never see their like again.
They crossed the finish line that Kintsugi and Woebot couldn't reach — and it still wasn't enough.
The Evidence Trap 🔗
So why did the companies that actually got FDA authorized still die?
Because not all evidence is the same. The evidence FDA needs for authorization and the evidence payers need for reimbursement are fundamentally different things. FDA evaluates controlled clinical studies — efficacy under ideal conditions, with selected populations, in structured settings. Payers want to see real-world, prospective, post-market evidence: does this actually work in practice, with real patients, in real clinical workflows, at scale? Does it reduce costs? Does it improve outcomes in the messy, uncontrolled world of actual healthcare delivery?
And here's the trap: if you burned your entire Series A and half your Series B getting through pre-market — running the clinical trials, funding the regulatory submissions, surviving years of FDA engagement — of course you don't have enough left over to generate the post-market evidence that payers actually need to see. You showed up to the reimbursement conversation empty-handed. Not because your product didn't work, but because you spent everything proving it worked under conditions that payers don't weigh as heavily.
This is the circular dependency that is strangling this entire sector:
FDA authorization needs pre-market evidence. Reimbursement needs post-market evidence. But post-market evidence needs FDA authorization.
And it gets worse. Reimbursement needs post-market evidence. Revenue needs reimbursement. Evidence generation is expensive, and the best way to fund it is with revenue. But you can't generate revenue without reimbursement, and you can't get reimbursement without evidence, and you can't collect evidence without authorization.
It's the classic chicken-or-the-egg bootstrap problem. By the time you've fed enough pre-market evidence into the front end of that loop to get authorized, you're out of capital to generate the post-market evidence that the back end demands. The companies that survived pre-market couldn't survive post-market. Not because their products failed — but because the system consumed all their resources before they could prove real-world value at scale.
How to Break the Cycle 🔗

But I think there is a solution. And it starts with both sides being honest.
I wrote an article recently about how the word "ideally" is a cop-out. It lets you recommend the cleanest, most expensive, least-likely-to-happen version of a solution — then walk away while the other side holds the bag.
Both sides of the regulatory table are doing this right now.
“Ideally”, for the manufacturer: give me the cheapest, fastest, smallest study possible. One and done. Get me authorized and never make me look at this data again.
But put yourself in FDA's shoes. A company comes to you with the bare minimum pre-market evidence and says "trust me, I'll collect more data after authorization." You know that once you open that gate, the incentive to invest in post-market evidence drops to near zero. History has shown this over and over. So FDA asks for more up front. And more. And more. Until the pre-market burden becomes the very thing that kills the company before they can file.
This is the stalemate. Both sides are saying "ideally" and neither side is making the hard call about what's actually achievable. Realistically? Neither side gets their ideal. And that's okay — but someone has to say it out loud.
Here's what manufacturers need to understand: FDA doesn't just authorize the sausage. What they're really authorizing is how the sausage is made. The 510(k) or De Novo is not a finish line — it's a statement that the company has demonstrated the discipline, the infrastructure, and the commitment to continuously ensure their device is safe and effective. That's what FDA is actually evaluating. Not just your clinical data. You.
So meet FDA halfway. Come to the table with an ongoing safety and efficacy commitment that's baked into your actual revenue model — not a vague post-market promise. If your business model requires the device to keep working for patients in order to generate revenue, you've just aligned your commercial incentives with FDA's safety mandate. That's not a burden. That's a competitive advantage. And it's exactly the kind of signal that gives FDA the confidence to let you through the gate with a lighter pre-market package, because they know you're not disappearing after authorization.
And here's the part that nobody talks about: post-market evidence isn't just cheaper to collect. It's actually better.
One of the biggest safety risks for AI medical devices is undetected overfitting — the algorithm performs beautifully on your clinical trial population but fails on patients who don't look like your training data. The other is data drift — the algorithm degrades over time because clinical workflows evolve, patient populations shift, and the real world moves on while your locked model stays frozen. Pre-market clinical studies, by design, cannot catch these problems. They're snapshots. Post-market evidence catches both. Continuously. In real time. On real patients.
So the math is clear: post-market evidence is cheaper to collect, it mitigates the exact safety risks that FDA cares about most, and it makes progress toward the reimbursement milestone — which generates revenue — which funds even more evidence collection. The flywheel starts spinning instead of collapsing.
I've argued that something closer to a 90/10 split — a focused pre-market package that also establishes the infrastructure for robust, continuous post-market evidence generation — gets the trade-off right. FDA gets ongoing assurance that the device is performing safely. Manufacturers get to market before they run out of money. And crucially — manufacturers would still have capital left to generate the real-world evidence that payers need for reimbursement.
I presented this framework at the FDA Digital Health Advisory Committee meeting in November.
The Race Nobody Has Won. Yet. 🔗
Right now, there are hundreds of companies building medical devices on generative AI — LLM-based therapy chatbots, autonomous clinical agents, multimodal models that read radiology images, pathology slides, and EHRs simultaneously, voice biomarker AI powered by foundation models, and generative clinical decision support.
FDA has cleared plenty of AI — but not an LLM-based, open-ended generative clinical agent. Not yet.
Out of the nearly 1,300 AI/ML medical devices the FDA has authorized — 295 in 2025 alone — the vast majority use traditional machine learning. The first foundation model only just crossed the finish line: Aidoc's CARE™ received FDA clearance in January 2026 for multi-condition CT triage, and a2z Radiology AI cleared a generalist triage system in November 2025. But these are vision-based imaging models with well-defined outputs — not the generative, language-based, non-deterministic AI that the next wave is building on. The generative AI frontier — therapy chatbots, autonomous clinical agents, LLM-based decision support — remains completely uncharted regulatory territory.
And here's why that should concern everyone building in this space: these technologies don't have a predicate. The 510(k) pathway — the route that 97% of AI medical devices use — requires you to show substantial equivalence to something that already exists. But nothing like this already exists. Which means most of these companies are looking at a De Novo pathway. The same pathway that took Kintsugi four years and $16 million and still wasn't enough. The same pathway that has a median timeline of five-plus years and costs around $5 million.
Consider the cautionary examples above. Those companies used traditional, well-understood AI, with established regulatory pathways, predicate devices, and decades of FDA precedent. And they still didn't survive. If you're building on generative AI, you don't even have that.
This is not a hypothetical problem. Google's Med-Gemini, OpenAI's ChatGPT Health, Anthropic's Claude for Healthcare — all explicitly disclaim: "not intended for diagnosis or treatment." Hippocratic AI, with $404 million raised and over 115 million patient interactions, carefully positions itself as "non-diagnostic." Nabla just partnered with Yann LeCun's new AI lab specifically to build "FDA-certifiable agentic AI" — future tense, because today it doesn't exist. Every single one of these is navigating around the regulatory void, not through it.
The question isn't whether foundation models will transform medicine — it's whether the regulatory system will let them get there before they run out of money trying.
The Regulatory Asymmetry 🔗
Meanwhile, thousands of unvalidated mental health apps operate freely under the "wellness" umbrella, making what are effectively clinical claims without clinical evidence. Patients can't tell the difference. The companies most committed to doing things right are the ones that cannot survive. The companies skirting the rules face no consequences.
I don't know what happened behind Kintsugi's closed doors. But I do know this: the same outcome will keep repeating until all sides stop saying “ideally” and start making the hard, constrained decisions together.
Start Here. Start Now. 🔗

I've outlined the problem. But I've also been publishing the building blocks of the solution — the actual tools, frameworks, and regulatory strategies for getting foundation model-based medical devices through FDA. If you want the deeper dive, start here:
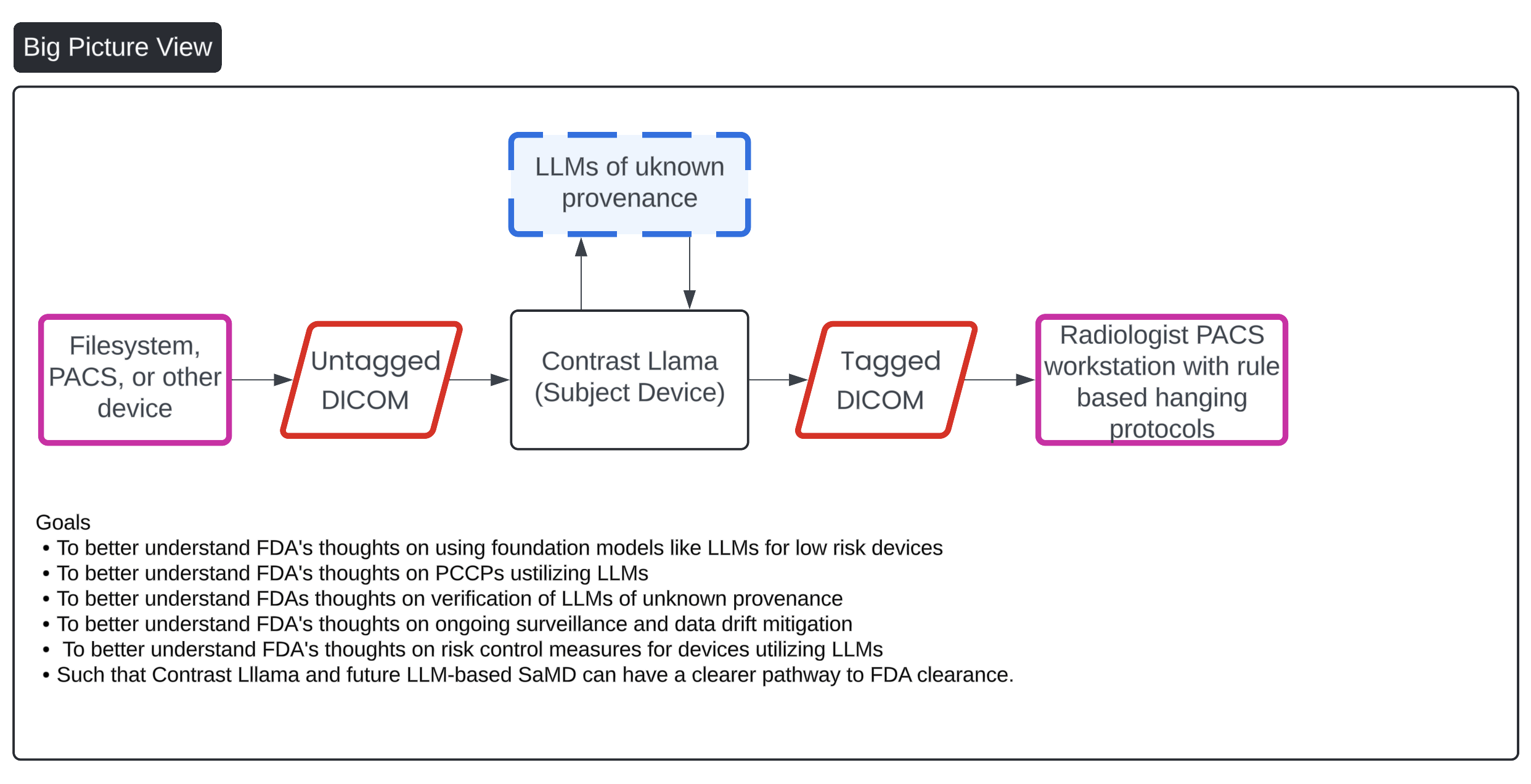
- How to get foundation models FDA authorized — best practices, FAQs, and a
real pre-submission we filed for our LLM-based device, Contrast Llama - A comprehensive look at the open-source radiology foundation models that are actually available to build on
These aren't theoretical. We filed presubs with FDA. We built AI/ML devices. We've contributed to 60+ AI/ML FDA authorizations and we presented these frameworks at the DHAC. And we've been publishing this thought leadership since 2024 — before the DHAC meeting, before the foundation model regulatory conversation went mainstream, and before most consultants in this space had even started thinking about it.
That matters, because here's the uncomfortable truth about choosing a consultant for this work: most regulatory consultants are very good at interpolation but not at extrapolation. They can look at what FDA has already done, find the closest precedent, and map your device onto an existing pathway. That works beautifully — when a precedent exists. But for foundation model-based medical devices, there is no precedent. There is no predicate. There is no prior De Novo to reverse-engineer. This hasn't been done before.
What you need is a team that can extrapolate — that can reason forward from first principles into territory that doesn't have a playbook yet. And that requires a combination of disciplines that is genuinely rare: deep clinical medicine, so you understand what the device actually does to patients. Deep AI engineering — not just "we use AI," but understanding context windows, anti-hallucination techniques, prompt architecture, model drift, and the difference between what an LLM can do and what it can be trusted to do. Decades of FDA experience, enough to have built an accurate mental model of how the agency thinks, what they'll push back on, and where they'll give ground. And real business experience — understanding funding constraints, reimbursement timelines, and the commercial realities that determine whether a company survives long enough to reach the market.
Without that last piece — the business lens — you end up with a consultant who keeps saying "ideally" and you're right back to the stalemate I described above. The whole point of this article is that "ideally" is killing companies. You need someone who can make the hard, constrained recommendation that accounts for what's actually achievable with the capital and timeline you have.
If you have the time, read the related articles — they're free, and they'll give you a head start that most companies in this space don't have. If you don't have the time, or if you're staring down a regulatory pathway right now and need someone who can extrapolate into the future, not just interpolate the past — reach out to me and my team at Innolitics.