
Introduction 🔗
You might be surprised to learn that the FDA wants to understand how your algorithm works to determine if you meet their burden of proof. This makes sense upon closer examination—a straightforward algorithm presents fewer risks and potential hidden bugs compared to a more complex one.
Some algorithms, like convolutional neural networks, have been in use for years, making their safety profiles, failure modes, and potential vulnerabilities well understood. However, newer technologies such as large language models are still being explored in terms of these factors. Therefore, providing a clear description of your machine learning algorithm or algorithm-driven software medical device is crucial for the FDA to offer valuable feedback during a pre-submission. This approach can help avoid unnecessary delays, rejections, or complications in the final submission.
I'll share a method I use to visually communicate key information to the FDA, simplifying their job and, consequently, yours. It's beneficial to work with a consultant who can guide you through this algorithm decomposition process. Ideally, they should have expertise in data science or machine learning engineering.
I've witnessed numerous FDA pre-submission meetings where the agency provided limited advice because they lacked a thorough understanding of the algorithm's engineering and scientific details. This understanding is crucial for assessing the level of proof needed for safety and efficacy. As a result, 60 days of waiting often went to waste. However, this doesn't have to be the case, and I hope to help you avoid such pitfalls.
Overview 🔗
The following diagram provides an overview of symbols used in subsequent illustrations, serving as a legend for easy interpretation. While this approach may not align perfectly with a data science perspective, it's designed to effectively communicate regulatory risk to the FDA. This method directs their attention to areas of concern, rather than requiring them to sift through extensive documentation. Such focused presentation can help avoid unsatisfactory pre-submissions or unexpectedly lengthy 510(k) application review processes.
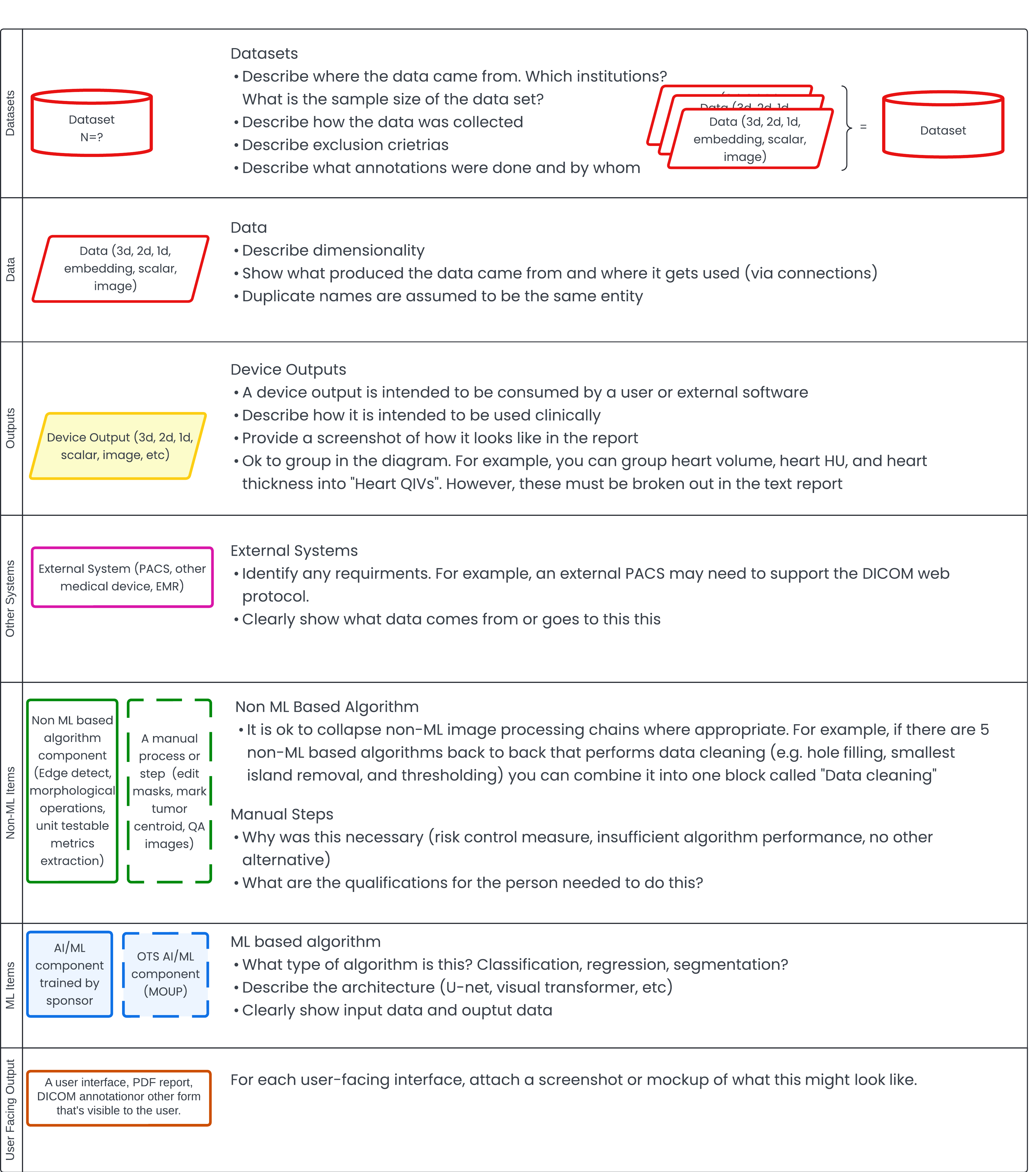
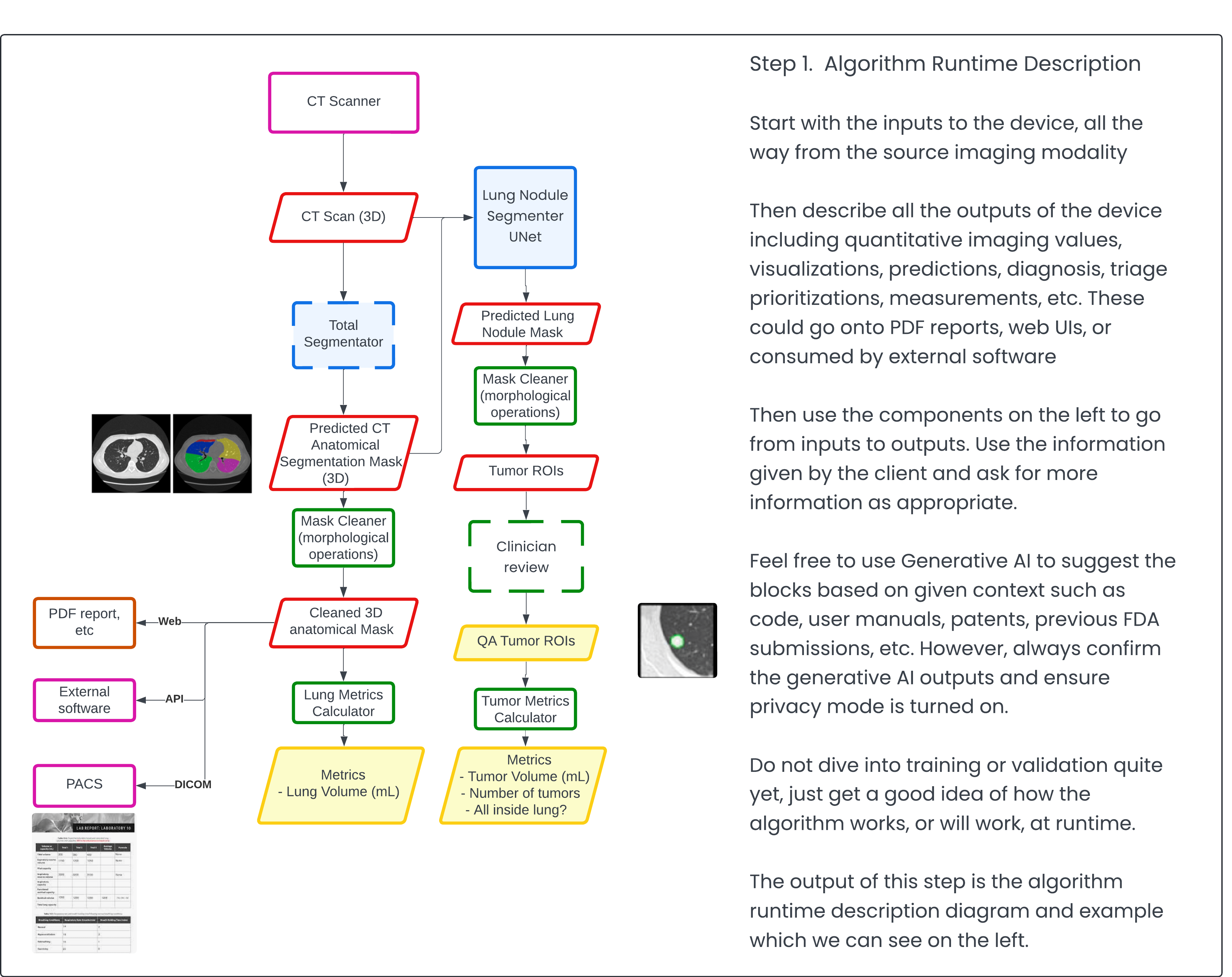
Step 1: Algorithm Runtime Description 🔗
Often, clients approach me with an already developed algorithm. In such cases, it's straightforward to reconstruct and describe how the algorithm works or is expected to function during inference and/or runtime. I prefer using a visual format (as shown below) because, as they say, a picture is worth a thousand words. I've deliberately color-coded the blocks to highlight areas where I believe the FDA should focus their attention. This approach reduces the cognitive burden on the reviewer and potentially puts you in the FDA's good graces.
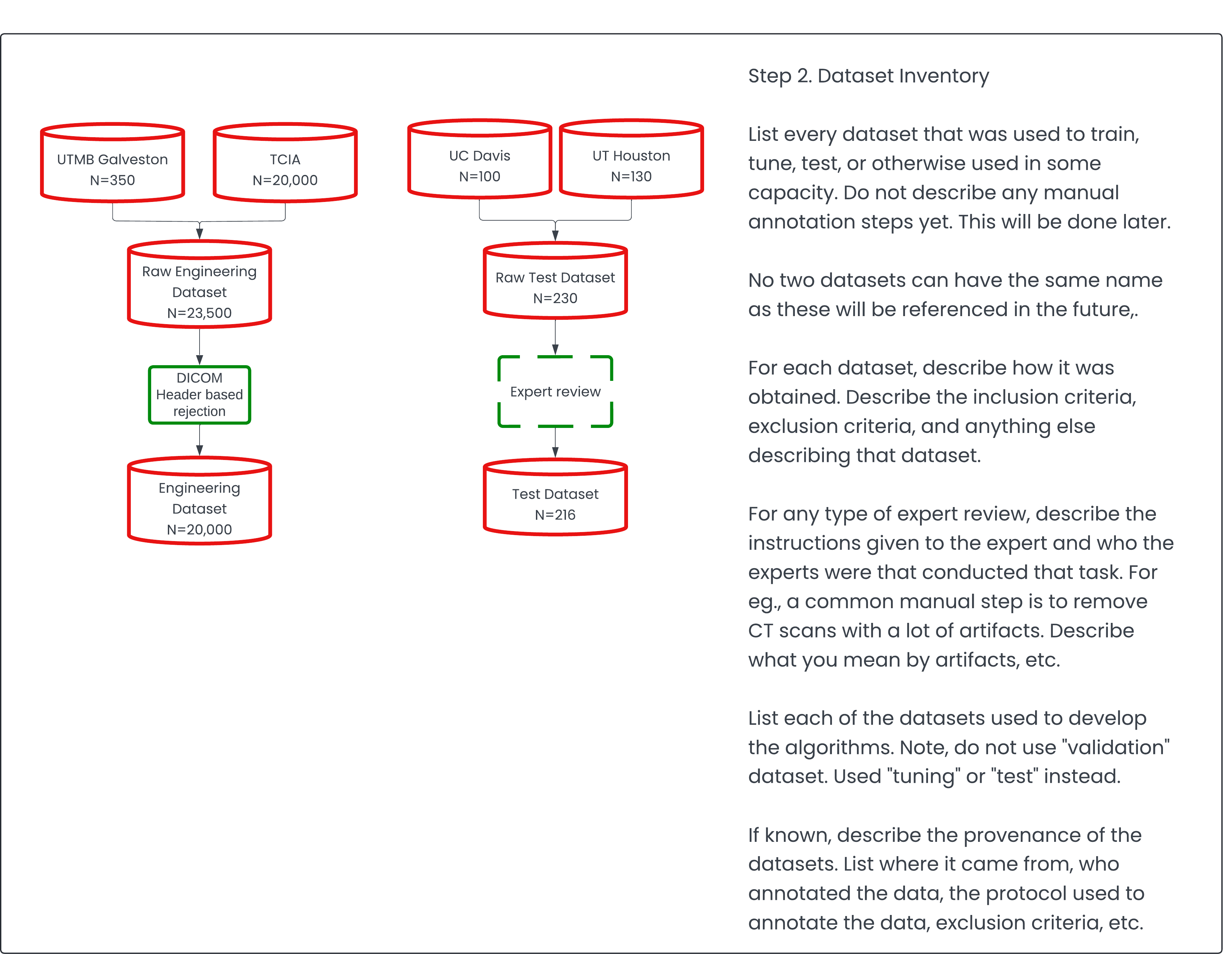
Step 2. Datasets Description 🔗
Typically, clients approach us with data they've already collected for algorithm training. In some cases, they may even have data suitable for an FDA performance assessment study, though they might not label it as such. Establishing a baseline of the client's existing data is crucial, as it can significantly reduce the time and effort required for additional data collection if they already possess suitable datasets.
Step 3: Non-ML Verification Testing 🔗
Did you know that unit tests can be sufficient for certain types of algorithm pipeline components? It's true. When you think about it, it makes sense. Some operations are so trivial, deterministic, and well-understood that they carry no inherent risks. For example, an edge detection filter is mathematically simple, well-understood, and intuitive—unlike a neural network with millions, billions, or even trillions of parameters. Therefore, the burden of proof must scale with complexity. In the simplest cases, even a sample size of one is sufficient to prove that a non-machine learning based algorithm is working as intended.

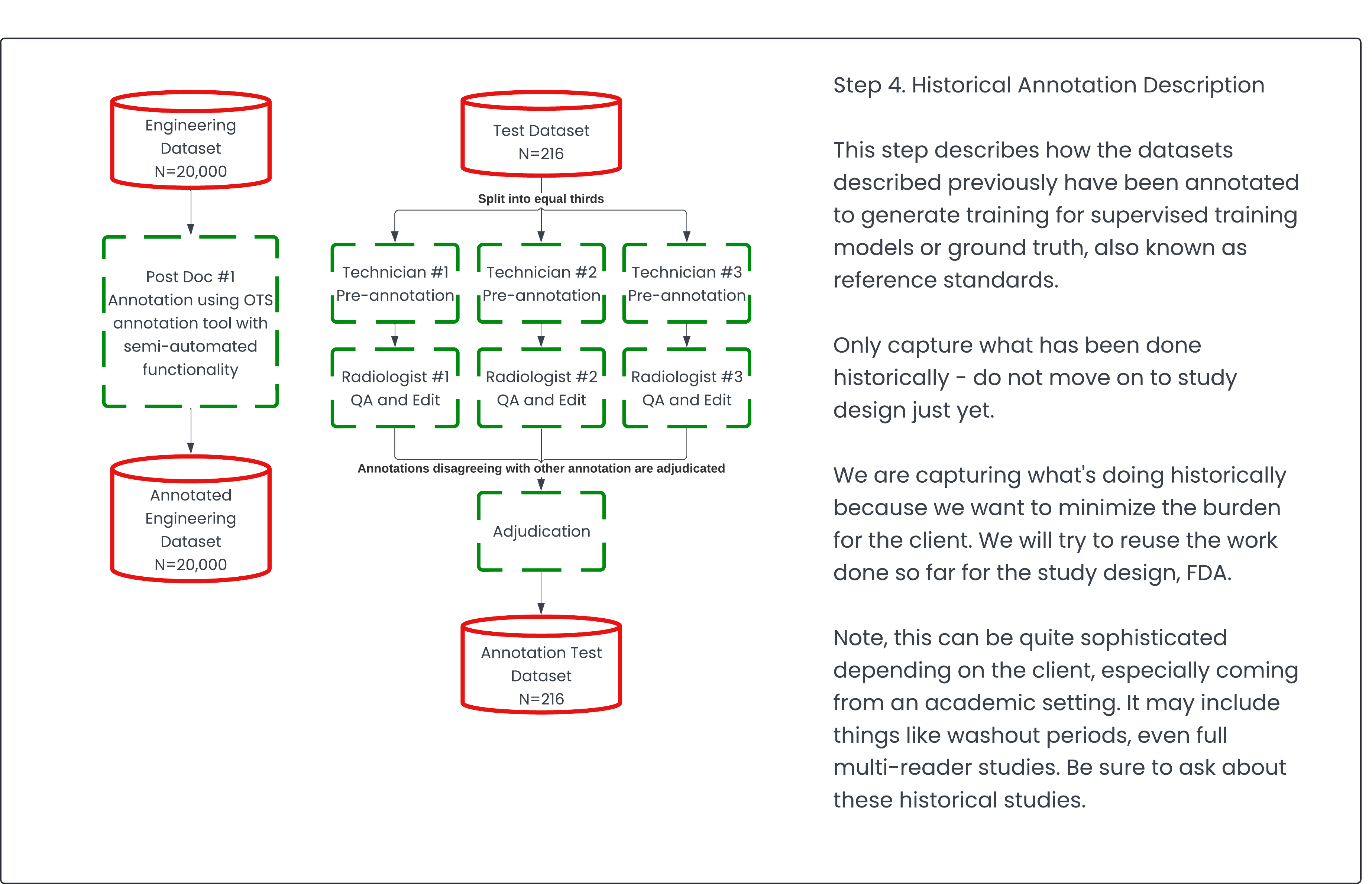
Step 4: Historical Annotation Description 🔗
When clients approach me, they typically have existing data along with some annotations. Most machine learning techniques still require supervision (a supervised learning strategy). We're quite far from achieving a completely unsupervised approach, though we're inching closer with foundation models.
It's crucial to document any annotated datasets that could be valuable for the FDA validation study. This documentation is important because the validation study is a significant cost driver in the subsequent 510(k) submission application.
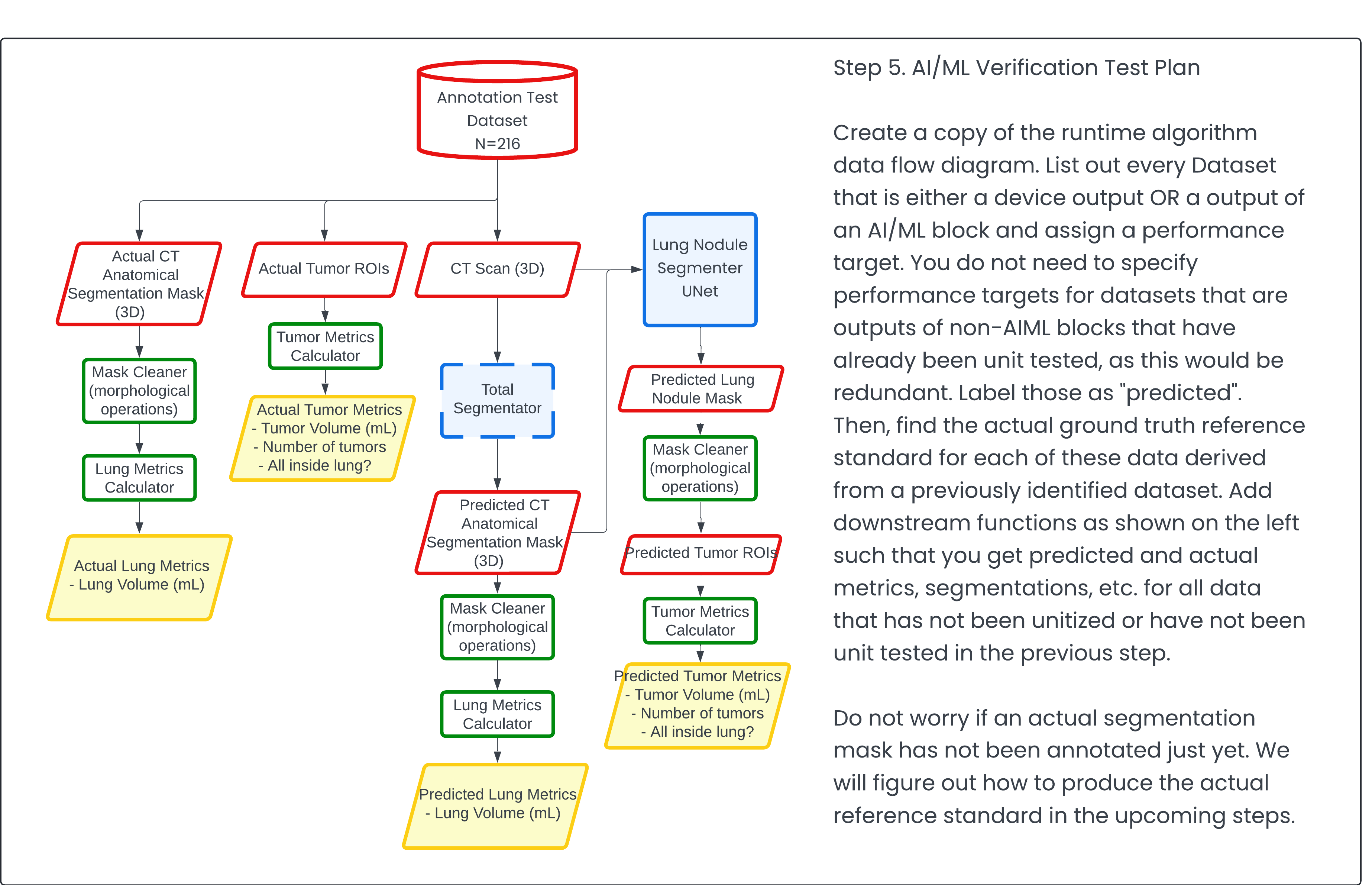
Step 5: AI/ML Verification Test Plan 🔗
A lot of consultants go straight to this part without giving FDA sufficient background to paint the picture to review. As mentioned before, without a strong grasp of how the algorithm works, how can FDA gauge the burden of proof required to overcome the safety and efficacy hurdle?
To overcome this hurdle, you need to provide the FDA with all the details upfront. This requires an engineering perspective, which I hope this article will elucidate. Once the mental framework is established, this step becomes straightforward. Think of it as verification testing—but for machine learning and AI components where you'll never be able to definitively prove that it's going to work in all cases or that it will generalize outside of your test set. However, you can prove it beyond a reasonable doubt that it will.
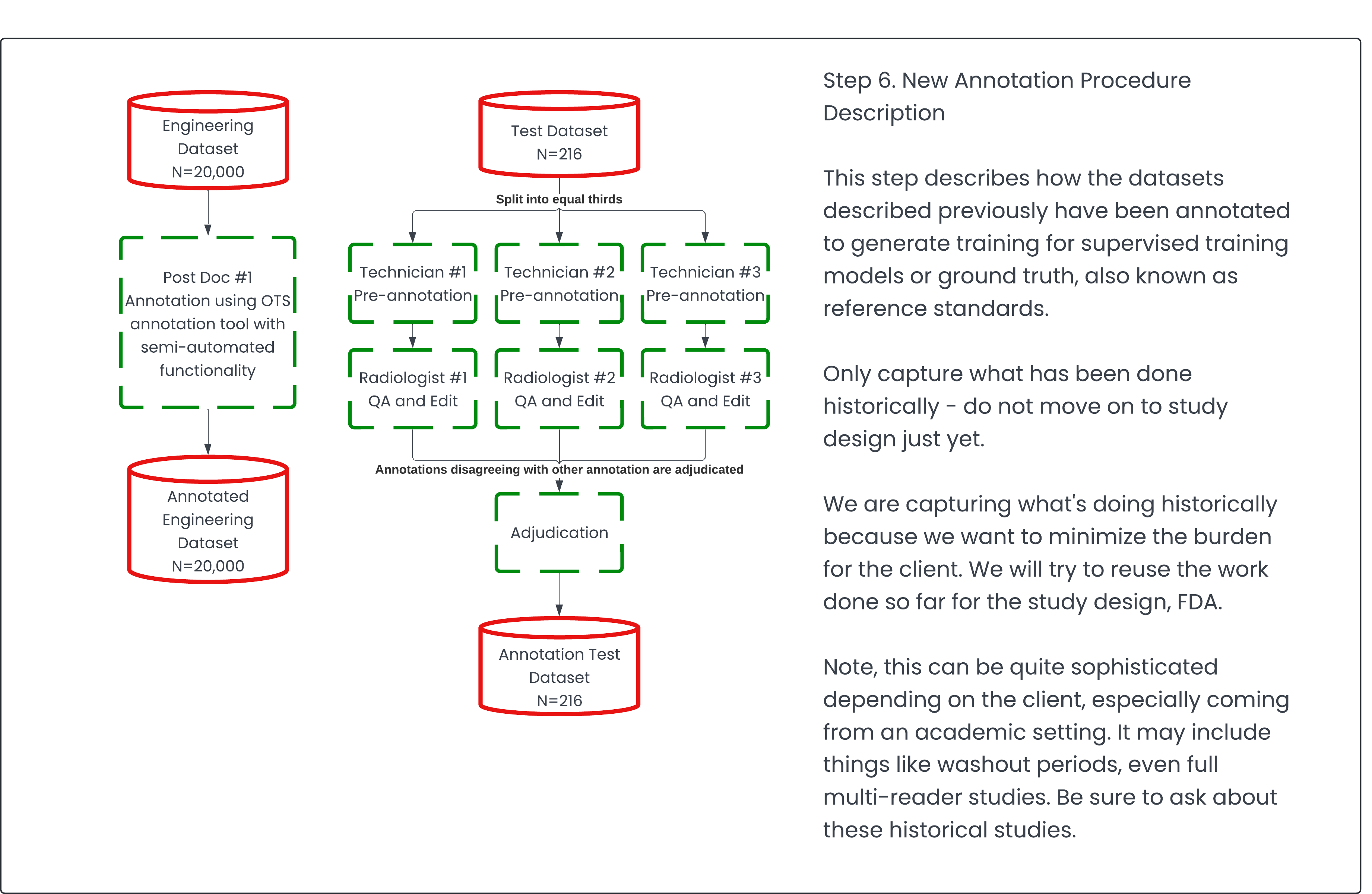
Step 6: New Annotation Procedure Description 🔗
This is where significant cost implications often arise, making it crucial to minimize the burden on the client. The process involves identifying the requirements needed to validate the functionality of the "black boxes" (as outlined in step 5), then determining the gap between these requirements and the existing data (from step 4). This gap represents what needs to be newly annotated or collected.
Data acquisition can be expensive, with costs ranging from $50 to $500 per CT scan, depending on the complexity of obtaining such scans. Therefore, it's vital to keep these costs as low as possible by leveraging existing data from previous steps. Moreover, annotation is another costly aspect, with skilled board-certified US radiologists charging between $300 and $800 per hour for their expertise.
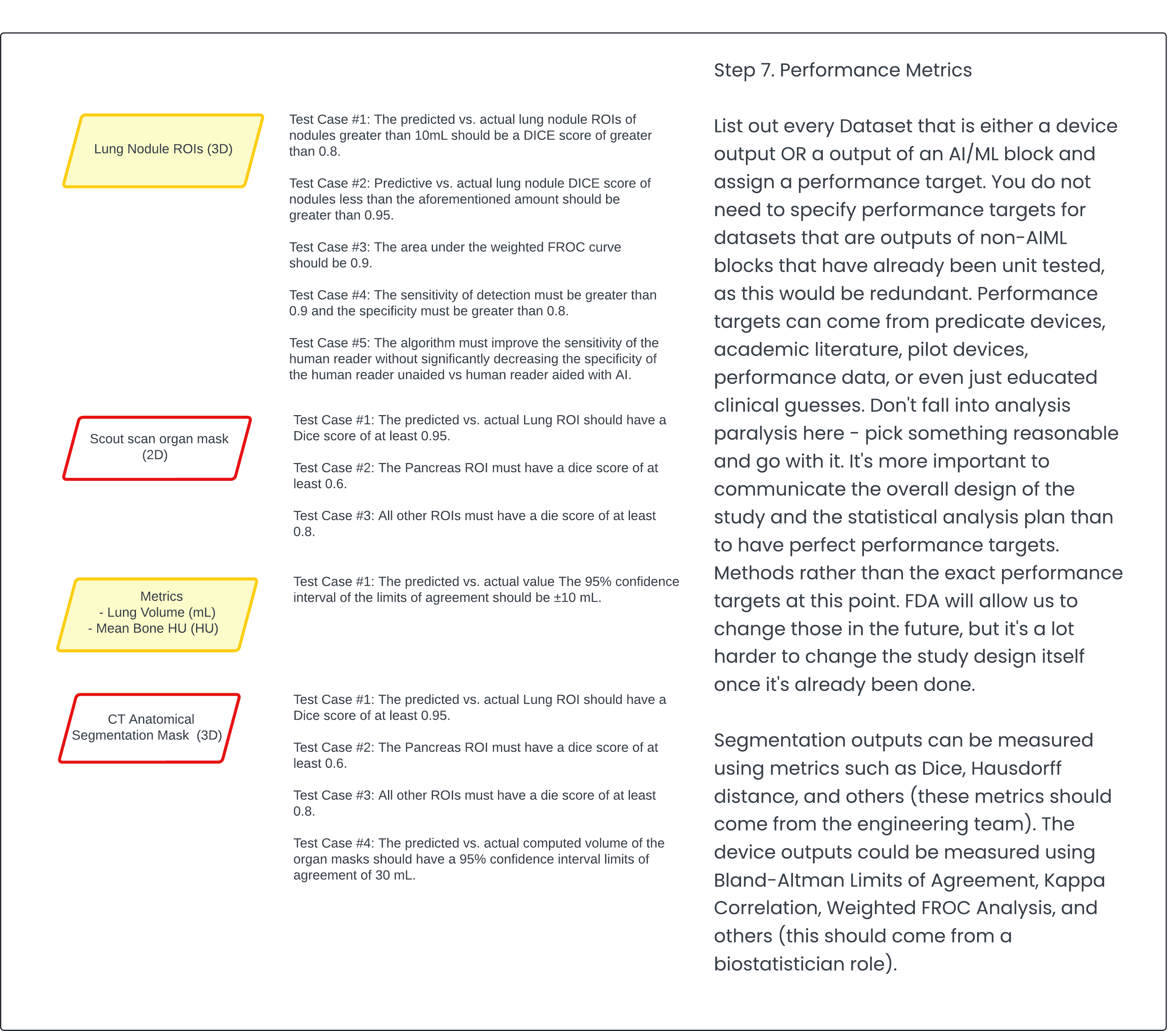
Step 7: Performance Metrics 🔗
Finally, set performance metrics and write them as software requirements. Capture the clinical performance assessment as a verification test. In my opinion, it maps better to a verification test because it is testing a requirement.
Conclusion 🔗
I hope you've gained valuable insights from this article. While this approach represents my personal perspective and methodology, it's important to note that it's not the only viable solution. However, it has consistently yielded positive results in my experience. I acknowledge my potential bias as a machine learning engineer, data scientist, and medical doctor, which may result in a more technical and engineering-focused approach. Despite this, I've found that the FDA responds favorably to this level of detail, likely due to their own background as scientists, engineers, and physicians.
The framework outlined above should equip you with the tools to prepare a successful FDA pre-submission, paving the way for your eventual 510(k) or De Novo application. However, if time constraints are a concern and you can't risk an FDA rejection, we're here to assist. Consider our AI/ML SaMD regulatory strategy service—just book a call to learn more.
Revision History 🔗
| Date | Changes |
|---|---|
| 2024-09-02 | Initial Version |
