Is this for me? 🔗
You will find this content useful if:
- You are developing a medical device that contains software (SaMD or SiMD)
- You are looking for concrete step-by-step guide and examples but all you can find are generic templates, non-definitive guidance documents, and ISO standards seemingly written in a foreign English dialect
- You would like a no BS formula for preparing a 510k in as little time as possible
- You care more about getting FDA approval rather than learning the theory behind it
- You have already written your software and need to submit an FDA application
- You are in the process of writing your software or have not started and are wondering when to start documenting
- Your device has AI/ML and you are wondering how to properly document this for the FDA
- You are frustrated by regulatory documentation not feeling worth it or adding much value to your organization
- You value solid examples as a learning tool
- You value clarity and practicality over learning the regulations from first principles
- You are an ethical human being and would only release devices you would feel comfortable using on your loved ones.
This is not for 🔗
You should probably pass on reading this if:
- Your device has no software
- You don’t learn much from examples and would rather learn the material from first principles
- You want to take shortcuts that compromise patient safety
Caveats 🔗
- There are no simple answer to regulatory questions. The answer is almost always “it depends.” The advice here is just means and averages but your situation may be an outlier. While this guide will prevent you from making some common pitfalls, it has some simplifications that may not always apply.
- Not all regulatory professionals will agree with our opinions in this article. In fact, some of them can stir up controversy. These are my opinions that have been validated through several FDA approvals, however, it does not mean they are “right” and it does not mean other approaches are “wrong.” There are several solutions to the question.
Why should I read this vs other content? 🔗
There are a ton of great materials about how to prepare your FDA application. Unfortunately, they don’t give you concrete advice and show you examples from a real FDA cleared application. This article will show you examples from K232613, a recently cleared AI/ML SaMD application that Innolitics built, validated, and got FDA cleared in just a year. If you learn from examples, this approach is for you.
When should I start documenting software? 🔗
While most consultants will tell you to start design controls as soon as possible, I think the answer is not as simple when you consider the bigger picture. It depends.
If you are a large company with an existing quality management system and products, you should follow your existing process—which usually means you need to start from the beginning.
If you are a startup making your first product without any existing framework, our recommendation—which is contrarian to most regulatory consultants—is to focus on the product market fit first. This typically means doing some early prototyping and customer interviews to ensure you have a product that people will buy (i.e. reimbursement strategy) and have a product that is technologically possible (i.e. feasibility analysis). Think about this. Which situation would you rather be in?
- I have an FDA approved product that does not help patients or physicians, and nobody wants to buy
- Physicians are clamoring to install my software in their hospitals, and I have several sales orders ready to sign pending FDA clearance
What should I consider early in development? 🔗
If you choose to focus on the product market fit first, please do not make the following costly mistakes:
- If you are using AI, make sure 50% of your data comes from the US. Ensure a US board certified clinician at least QAs your data. Also, you may be requested by FDA to provide the qualifications of your Ground Truthers so have this information available.
- If you are using AI, make sure your training and test set comes from different sites.
- If you are using AI, make sure your training data annotation protocol is well document and is the same one used for testing.
- If you need to do a clinical performance assessment, do a pre-submission first
- Ensure your reference data is at least reviewed by qualified US board certified physicians with 5 years of post residency experience.
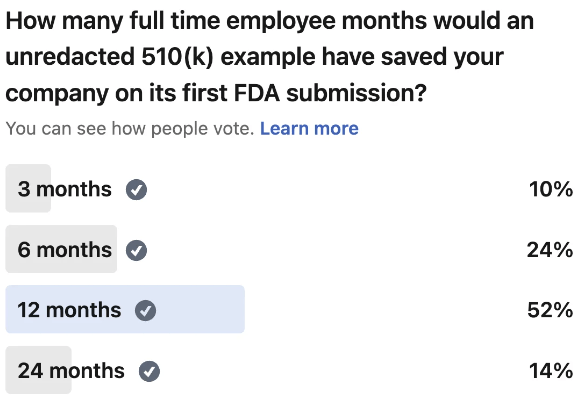
- I see too many clients come to me with no budget remaining for the FDA application. Or they assume it the whole thing just costs the $6k you have to pay the FDA. The submission fee is insignificant when you account for the labor involved in preparing the documentation. It can take your team 6 to 24 full time employee months of effort to prepare the FDA application. Ensure you have enough runway to absorb this cost. So if your average employee costs you $120k, ensure you have at least $60k to $240k budgeted for the FDA submission. The last thing you want is to do is raise an unexpected down round.
- Have realistic expectations about AI. You may need to do another round of R&D during the 510k preparation process because your algorithms may not perform as well in validation as they do in development. If it took you 12 months to develop the algorithm, consider adding a 3 months buffer to fund another round of R&D to fix the issues found during validation.
The Pre-Submission Meeting 🔗
Requirements 🔗

- Imagine you are planning a vacation. Your primary focus is to be at specific locations at predetermined times, while the events in between hold less significance. You wouldn't meticulously plan the exact taxi driver you'll use or whether you'll turn left at a particular street or walk an extra block, as these details are inconsequential to your objectives. Your explicit planning revolves around aspects that would bring joy to your partner, mitigate potential risks, or substantially contribute to the overall purpose of your vacation. Similarly, software requirements should document behaviors that are crucial to the system. For this reason, I prefer to conceptualize requirements as a form of design constraint. Some examples include:
- Mitigate a safety risk
- Mitigate a user experience risk
- Mitigate a business reputation risk
- Mitigate a cybersecurity risk
- Requirements must be unambiguous and verifiable. Requirements become your specifications.
- Requirements must not conflict with other requirements.
- As a general rule of thumb, most of my SaMD projects end up with around 100 requirements. This is not a perfect measurement, but 1000 requirements for a relatively simple SaaS SaMD is probably overdoing it.
- The number of requirements may increase or decrease over time. It is easier to add new requirements than to remove existing ones so keep it simple to begin with.
- You don’t need to get all of them upfront. It is ok to add requirements after the product is released and you get feedback from users. You may have software behavior that you did not know was important until a customer complains about it. Don’t beat yourself up or penalize your team for that. For example, padding and spacing around buttons is usually not something I write a requirement about. However, a customer may complain that text is cut off in their non-english translation. Then it becomes something worth pinning down as a requirement.
- The easiest way to calibrate yourself on how detailed requirements need to be are to look at a couple of examples.
- All requirements must have at least one verification activity tied to them.
Risk Management 🔗
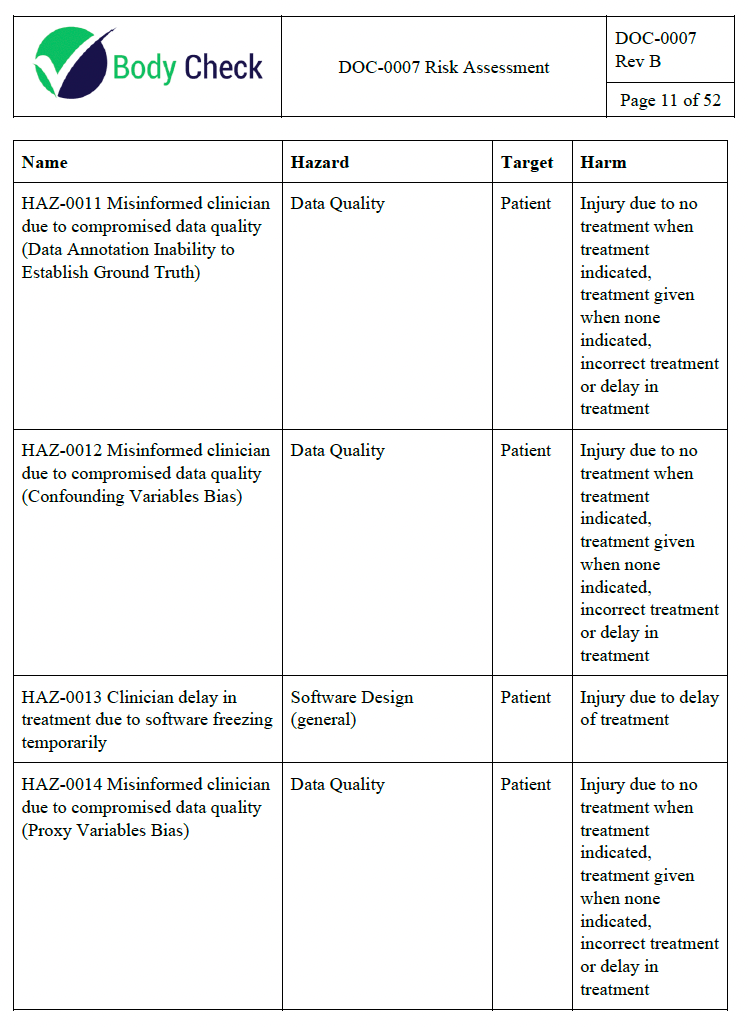
The first time I did this on my own I ended up wasting 6 months of cumulative full time employee effort by getting way too detailed than was necessary. The second time I did it I ended up with all my risks being severe harm with low probability which resulted in the product almost being recalled due to suboptimal risk evaluation. The fourth time I did it was a little better. 25+ times later, I have a couple of tips to avoid some serious pitfalls.
- Start with the clinical picture. Ask your clinical team how they can envision the device leading to patient harm.
- Use “misinformed clinician leads to suboptimal care” as most of your endpoints. Do not exhaustively list all the ways the practice of medicine can lead to harm unless your application calls for it. For example, if you are creating a radiotherapy treatment planning app, you should be considering how radiation burns can arise out of the usage of your software. However, if you are detecting the cancer, you don’t need to worry about how your software can be involved in a sequence of events leading to radiation burns. You can just stop your risk analysis at “misinformed clinician leads to suboptimal care”, which could lead to a lot of harms but you don’t need to worry about it unless you have precedent for such a scenario happening in the past.
- Next, your engineering and product team should be involved in brainstorming ways your device could lead to a misinformed clinician. This could be due to a defect leading to an incorrect result leading to a misinformed clinician. It can even be due to a misleading report or UI leading to a misinformed clinician, even if the device is working as intended. As an aside, this is why the failure mode effects analysis (FMEA) is not sufficient as a risk analysis methodology on its own—even devices working without failures have risk.
- Calibrate your team on your risk and probability. A common trap is to attempt to document every single scenario no matter how unlikely. Then you will be stuck with a bunch of useless risks that sap your team’s resources from handling the ones that actually matter. Use your judgement and if a risk feels trivial or extremely unlikely, it probably is. Don’t clutter your risk management file with hundreds of impossible risks and miss the one that actually causes patient harm.
Verification and Validation 🔗
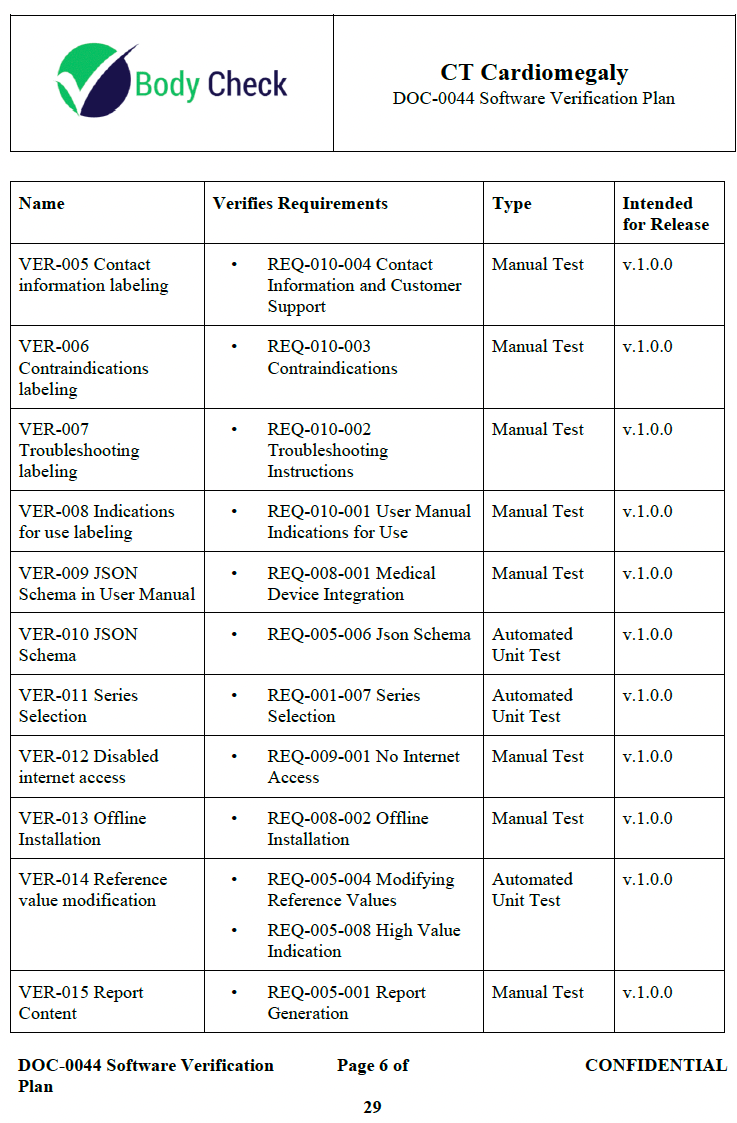
There are several traps that teams can fall into.
- Over-reliance on automated tests. Do you really need to write a browser based automation test just to ensure a button is a shade of red? A manual test—though less sexy from an engineering standpoint—can be easier to write, update, and debug.
- Over-reliance on tests. Did you know there are other verification activities other than just tests? You can do independent code inspection to verify a requirement is met.
- Over-documenting tests. Did you know that not all tests need to have a requirement tied to them? It is true. A lot of automated tests actually fall into this category.
- Over-rigid test schema. Tests do not need a one to one relationship with requirements. A test can verify multiple requirements. However, you don’t want one test that verifies every requirement either. See the example video for some tips on where to draw the line.
- A plan for the plan for the plan. The FDA cares less about the “why” and more about the “how” and “what” was tested. I often see teams making it way harder on themselves than necessary by bloating their V&V plan with too much complication that the team is not following anyway. Less is more. Your verification plan can just be the test steps you plan to execute. This often happens because the team uses templates that are more appropriate for teams 100x their size.
- Over-siloed. There is a common belief that engineers cannot be involved at all in verification as if there needs to be a great firewall between QA and engineering. While the merits of this practice make sense, in practice it is detrimental for all but the most highest risk applications. Engineers test their own code all the time in a practice called “test driven development.” This has shown to create better software, not worse.
- Deletion dilemma. Sometimes you will have that one user need or requirement that is not adding much value. This typically only becomes a problem during V&V planning. Just delete it, it never belonged in the first place.
User Needs 🔗
- User needs should start with “User needs to…”
- They should represent what the user wants out of the software
- They are typically written by the product and clinical team, whereas requirements are written by product and engineering team
- All user needs must have at least one validation test tied to them
- It is not required to submit user needs as part of the 510k submission but it is an important part to explore product market fit. Don’t you think it is useful to have a well researched list of needs your users want?
Potato vs Potato 🔗
Sometimes you will find user needs that could be requirements, verification tests that could be validation tests, hazardous situations that could be the last step in the sequence of events, hazards that could be harms, and vice versa. I have wasted months of my time debating the subtle differences between the two. My advise is to pick a side and move on or include the offending topic in both camps if your team cannot come to a consensus. It is not the end of the world if a verification test was actually booked as a validation test or if you have slightly duplicative V&V tests. The important part is that the activity gets done and you save your time to actually make the product better, safer, more accurate rather than having a scholarly debate about nuances.
But One More Feature… 🔗
Here is a story. A large medical device client was almost ready to submit but they kept delaying because of one more feature. The worry was that it would be hard to add new features once the application was submitted. This is not true. You can add features without having to resubmit a 510(k). Also, resubmitting another 510(k) is not as painful as it sounds if you use the right tooling. Many of the documents that you will access to, can also be used for a Special 510(k), which is quick and “light”. There is a misconception that your device needs to be completely “done” before submitting to FDA. A software product is never “done”. There is ongoing maintenance, new features to add, and new bugs to fix. Software in a medical device or software as a medical device is no different. Don’t fall into the “but one more feature” trap. In my opinion, you should pick a release date and try to stick with it unless there are serious issues that would lead to patient or business repetitional harm. And don’t forget the power of the underutilized Special 510(k) as a regulatory strategy.
KPI Wars 🔗
A key performance indicator (KPI) for the regulatory team could be “zero FDA submission findings.” While this may seem reasonable at first, it actually sets up the organization for failure. Getting zero FDA findings on a submission is almost impossible and it would misalign the regulatory and company wide objectives. The company should get the device out into the hands of users as quickly as possible while maintaining safety and efficacy. Sometimes this means taking strategic risks on the FDA submission that would require a trusting relationship between the C suite and the regulatory team.
I think there should be an expectation to get some findings but not too many that you are unable to address them all in the 180 days. The FDA almost always sends a AINN (aka FDA hold) letter and you have 180 days to address their concerns before the application is automatically withdrawn.
Another suboptimal KPI is “100% code coverage or 100% testing automation.” This results in chasing numbers rather than optimizing quality.
Frequently Asked Questions 🔗
Need Help? 🔗
Do you need a partner that can either guide you through the process or just take it off your hands completely?
Revision History 🔗
| Date | Changes |
|---|---|
| 2024-05-22 | Initial Version |