

Introduction 🔗
Some of the activities FDA asks us to do are a waste of time. Don’t get me wrong, the FDA does a great job (thank you!) but regulating the large and evolving range of medical devices is difficult. The regulations won’t always make sense.
Some of the activities your company’s SOPs require won’t make sense, either. At smaller companies with a new QMS, these pointless activities are holdovers from templates that were copied without a clear understanding of their applicability. At larger companies, it may be because the SOPs have grown bulky from being patched by the quality team by miscellaneous findings from audits across the globe and different product lines.
Regardless of the reason, it’s frustrating to work on something that feels pointless. Not only is it a waste of resources, but it also erodes a sense of purpose. Most of us don’t like wasting our time on pointless activities, even if we’re being paid to do them.
If you’ve ever been working on a medical device and thought, “this is a waste of time,” this article is for you. It will provide a framework for understanding which activities are necessary and how to proceed if you believe one isn’t.
Pursue the Why 🔗
If you’re following a procedure and it feels like a waste of time, you should ask: “Why are we doing this?” Sometimes, once you dig into it, there is a good explanation that you hadn’t thought of (like Chesterton’s Fence). Once you know it, you may not enjoy doing the activity, but at least you’ll see why it’s necessary. Other times, it really may be a waste of time. Once you’ve identified this, you can act appropriately. You may be able to change the procedure, or, at least, you’ll know to complete it doing the minimal amount of work necessary before moving on.
One of our values at Innolitics is pursuing the why. It’s a key reason we can help medtech companies create and clear their software so quickly. It’s tempting to settle for the status quo, but pursuing the why can lead to 10x-efficiency improvements and it fosters ownership and engagement.
Despite our values, we must admit: we don’t always need to pursue the why. Parents of three-year-olds will agree. Why? Why? Why? At some point we break: Just do it! Trust me. I don’t have time to explain, we need to get to school.
The same is true at work. Not everyone doing a task needs to know why they’re doing it. If a task can be carried out by following a clear set of instructions, you don’t need to know why the instructions were written the way they were. Some cooks just follow recipes. They make amazing meals without knowing why the steak was seared or the flour was sifted.
Not only does everyone not need to know why, but many people don’t care. They’re content following the procedure and getting the work done, especially for tasks outside their circle of excellence. Engineers may not care to explore the distinction between user needs and design inputs. Regulatory professionals may not care why Python is sometimes better than C++. Pursuing the why can be an abstract, boring, pedantic or philosophical distraction.
Understanding “why” is a deeper level of knowledge that only becomes necessary in the following situations. First, if you’re creating a new recipe. Second, if you’re following a bad recipe and you need to adapt on the fly. Third, if you’re doing something that can’t be fully captured by a recipe. In these cases, knowing the why is important.
Software systems are built in layers. Often, the engineers working on a higher layer won’t need to understand the underlying layers. However, sometimes things will go wrong in a lower layer. This is called a “leaky abstraction.” When there’s a leak, you need to dig in to get a deeper level of understanding so you can fix the problem.
In what follows, I’ll discuss how to go about pursuing the why within the context of medical devices.
Medical Device Stakeholders 🔗
When pursuing the why, you need to keep in mind that medical devices have many stakeholders. The needs of these different stakeholders often overlap, but some have unique needs. For example, an AI/ML algorithm for MRIs may have the following stakeholders:
| Stakeholder | Key Needs |
|---|---|
| Patients | To have their medical issues properly diagnosed |
| Radiologists | To be able to quickly interpret the generated reports |
| Food and Drug Administration (FDA) | To ensure medical-devices are safe and effective, to minimize review costs, to provide a consistent process |
| Health and Human Services (HHS) | To protect private health information by enforcing HIPAA |
| MRI Technicians | To efficiently learn and run the relevant MR sequences |
| IT Departments | To be able to easily and securely deploy and maintain the software |
| Manufacturer | To protect intellectual property, to minimize costs, to have a highly marketable and reimbursable device |
| Insurers | To understand the risks and benefits of new technology so they can reimburse efficiently |
If something seems like a waste of time, it may be because there’s a stakeholder with needs you haven’t considered. Thus, it’s important to be sure you’re aware of the various stakeholders. Once you know the needs of these different stakeholders, you can prioritize. For the rest of this article, I’m going to focus on two of the most important stakeholders: the FDA and Patients. This scenario can be illustrated as follows:
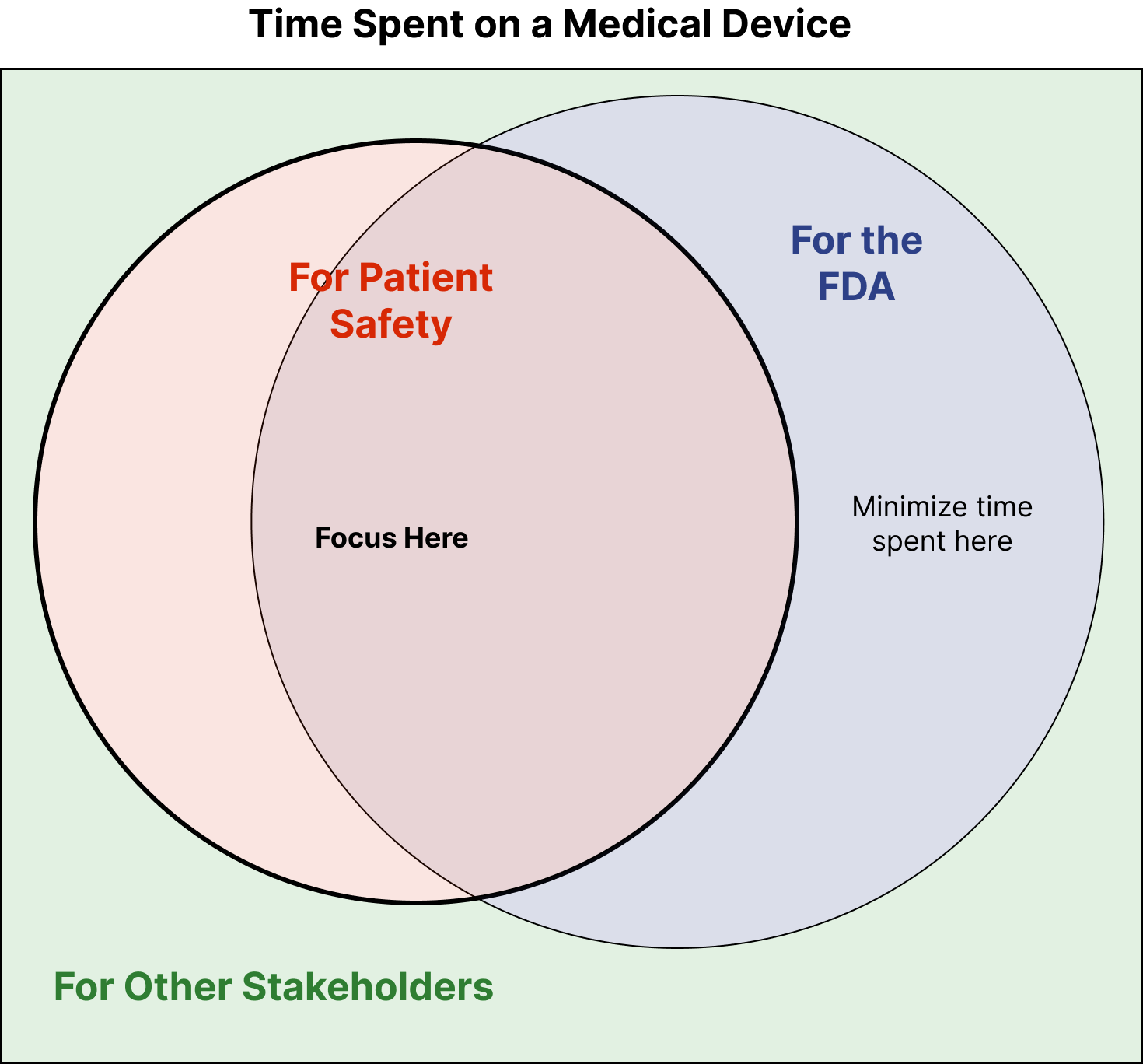
Each circle represents the time spent meeting the needs of that stakeholder. The circles overlap because some activities, e.g., Safety Risk Analysis, help meet the needs of both the FDA and Patients.
The circles overlap a lot, but some activities are just “checking the box” for the FDA and don’t make the device safer or more effective. This is the area that’s in the blue circle but outside of the orange circle. When we pursue the why we’re able to identify these activities and minimize the time we spend on them.
Of course, much of what we do for the FDA does make the devices safer. This is the overlapping region in the middle. Perhaps more surprisingly, there are also activities that should be done to make the device’s safer, which the FDA doesn’t require. (E.g., doing thorough code reviews for Software as a Medical Device.)
Then there is also time spent on activities that aren’t for the FDA or for patient safety. E.g., completing patent applications, which is done to meet the needs of the business.
More complex Venn diagrams can be created when there are multiple stakeholders, and it can be useful to understand where they overlap.
The FDA Has a Hard Job 🔗
Ideally, everything the FDA requested would be necessary and sufficient for making a safe device. I.e., the orange and blue circles would perfectly overlap. To understand why this is impossible, it’s helpful to consider the FDA’s needs.
The FDA regulates a huge range of devices. What makes sense for a prostate surgery robot won’t make sense for a face mask. To address this, they have regulations for specific risk-tiers (Class I, II, or III) and special controls for certain product categories. They will also make comments such as “device cybersecurity design and documentation are expected to scale with the cybersecurity risk of that device” or “the level of confidence, and therefore the level of software validation, verification, and testing effort needed, will vary depending upon the safety risk posed by the device”. Nevertheless, there are still regulations that don’t feel like they apply to your device.
FDA must also balance being abstract or concrete. Concrete guidance is helpful since it makes it clear what you need to do. But the more concrete the guidance is, the more quickly it ages as the technology landscape changes. This is why FDA guidance doesn’t usually say: “Use Git to track your source code,” which most engineers would understand. Instead they say “Use robust software configuration and change management,” which many engineers won’t understand.
FDA also must keep their guidance relatively short and accessible. They need reviewers and auditors with varying ability and expertise to be able to administer it.
Given how difficult their task is, I think the FDA does a great job overall. (Thank you!) Even so, there are activities the FDA requests which are a “waste of time.” I like to keep in mind that it’s unavoidable, so as to avoid being frustrated or jaded.
Sources of Medical-Device Regulatory Requirements in the US 🔗
If you’re working on something that seems like a waste of time, and you want to know if you’re doing it for the FDA, then it’s helpful to know the sources of medical-device regulatory requirements in the US. It is often useful to understand the difference between the following:
| Item | Description | Example |
|---|---|---|
| Federal Statutes | Laws enacted by the US congress. They often delegate the specifics to a relevant federal agency, which then drafts the necessary regulations. You can read the United States Code here. | The main statute applying to medical devices in the US is the Federal Food, Drug, and Cosmetic Act (FD&C Act) and its subsequent amendments. The acts are re-numbered and entered into the United States Code (U.S.C.). |
| Regulations | Rules or directives made and maintained by a federal agency, based on statutes, to implement those statutes. They’re maintained and published in the Code of Federal Regulations (C.F.R). | Medical device regulations are encoded in Title 21, Chapter I, Subchapter H. Specific sections are referenced using a short hand like 21 CFR 820 (the Quality System Regulation). |
| Guidance | Documents that “represent the agency’s current thinking” on a topic. Unlike statutes and regulations, they are not legally binding, but in practice it’s easiest to follow them as if they were. | There are dozens of FDA guidance documents that apply to medical device software. The 2023 Content of Premarket Submissions for Device Software Functions is a big one. You can search the FDA database here. |
| Standards | Standards are developed by non-government organizations. They’re not legally binding unless a regulation specifically states it’s required. They provide an abstract list of requirements. | The primary standards that apply to medical device software are ISO 13485, IEC 62304 Software Process, ISO 14971 Risk Management, and ISO 62366-1. |
| Technical Information Reports (TIR) | Most organizations that create standards also publish TIRs. Their content may be similar to standards, or it may be more informational. Regardless, they undergo a less rigorous review and approval process. | The Association for the Advancement of Medical Instrumentation (AAMI) publishes useful TIRs. For example, “AAMI TIR45 Guidance on the use of AGILE practices in the development of medical device software” |
Most of the time the activity will trace back to a guidance document.
Is This A Waste of Time? 🔗
Reading the guidance and regulations is one thing, but it’s also important to know what FDA emphasizes in practice. E.g., one could read the Software Validation guidance and walk away thinking a lot of things are important that the FDA rarely, in practice, asks about. This is where regulatory professionals with experience in your domain can really come in handy. They know what FDA asks for for a certain type of device.
So, if you’re wondering whether an activity is a waste of time, the first thing to do is determine the relevant stakeholders. Who is requiring that we do this? If it’s the FDA, then the next step would be to identify the FDA guidance that describes the activity. In some cases, you may even need to consult the underlying regulations.
Once you’ve found the relevant guidance, read it carefully. Identify if the activity is required or if it’s recommended based on the risk-level of your device. If the latter, and if you can make a case against doing it, then document your thought process and move on.
If your company’s SOPs are stricter than the FDA guidance, then work with whoever’s responsible for your SOPs to change them. Remember that nobody wins when a team is bogged down with busywork— streamlining SOPs is in everyone’s best interest!
An Example 🔗
Greg is an engineer working on a new AI/ML-enabled SaMD for Medical Device Corp. The device will run as a docker container and is expected to run within an AI marketplace. The container doesn’t have any open ports. It reads its inputs from a locally mounted directory and writes its output files to a locally mounted directory. It does not persist any Protected Health Information (PHI).
As Greg is working through Medical Device Corp’s P-0057 Security Risk Management SOP, he reads: “All devices shall undergo annual penetration testing.”
Greg doesn’t feel that penetration testing is necessary given the security controls and the inherent safety of the design. He thus believe penetration testing would be a waste of resources.
Greg reviews the document’s revision history for the P-0057 to see if there are any notes that explain why the hard requirement for pen testing was included. There is no such note. Greg then asks the Quality Management Representative if they know why pen testing is a hard requirement. They don’t know.
Greg then decides to do some digging on his own. It would be easier to just do the pen testing and move on, however, he dislikes the idea of wasting resources on pointless testing.
Greg identifies the various stakeholders. (If he was lucky, the product definition and user needs document would list these, but he’s not lucky.) He spends some time asking the product owner and project manager who would care about pen testing. He arrives at the following list of possible stakeholders:
- The AI Marketplaces Hosting the Container
- The FDA
- The HHS (per HIPAA)
- Medical Device Corp
Greg gets in touch with the AI Marketplace that they will be deploying at, and confirms that they don’t require pen testing. He reads online that HIPAA doesn’t require pen testing. Finally, he confirms that his company, Medical Device Corp., doesn’t require pen testing for any other business needs. This leaves the FDA as the final stakeholder.
He identifies the relevant FDA guidance with a few online searches—Cybersecurity in Medical Devices: Quality System Considerations and Content of Premarket Submissions. He finds the section titled "Cybersecurity Testing” and identifies that the document says “FDA recommends that the following types of testing [including pen testing], among others, be considered for inclusion in the submission.”
After pursuing the why, he decides there’s not a good reason to include pen testing for this device. He thus submits a document change request to alter P-0057 to no longer make pen testing a hard requirement.
