
Key Takeaways 🔗
- Statistical Power Is Insufficient Alone: Statistical power does not prove generalizability for FDA clearance; addressing biases in data such as age, race, and device types is essential.
- Data Requirements for Testing: For non-CADx devices, approximately 200 subjects are recommended, with at least 50% from the U.S. (100% from U.S. is even better), and data should capture variability in demographics and device types.
- Handling Algorithm Changes: Retraining algorithms may not require a new 510(k) if changes are not significant, but adding new features or indications likely does.
- Setting Performance Targets: Justify performance targets based on predicates, literature, or pilot data; aim for AUC > 0.9 for diagnosis/detection and DICE > 0.8 for segmentation.
- Importance of Cross-Functional Teams: Involving engineering, clinical, marketing, and regulatory stakeholders ensures a balanced approach to product development.
- Strategic Pre-Submission Meetings: Schedule pre-sub meetings after algorithm prototyping to focus on high-risk areas like study design; clear communication with FDA is crucial.
- Risk Assessment Based on Confirmation Bias: FDA evaluates AI devices based on the confirmation bias introduced; devices influencing diagnosis or highlighting locations carry higher risk.
- Study Design for CAD Devices: Conduct standalone performance testing before multi-reader studies to identify issues early and avoid costly mistakes.
- Constraints on Generative AI Models: Generative AI devices have not been FDA-cleared; constrain inputs and outputs and prevent misuse to align with regulatory expectations.
- Automation in Biostatistics: Automate statistical analysis to efficiently handle subgroup analyses, which are often required by FDA feedback.
Slides 🔗























Transcript 🔗
Yujan: Good afternoon, everybody. Today's topic is about how to get AI/ML software as a medical device FDA cleared. My name is Yujan Shrestha. Thanks for being here today. First of all, thanks to my wife and family. This time when I get back, I'll also change some dirty diapers too.
Yujan: My team is named Analytics. We are 18 strong software engineers and regulatory consultants. Here are some of our services. Our company offers software engineering services and also regulatory services. If you want to know more, our booth is right over there. You can see it from the auditorium. After this presentation, we'll be happy to take any questions at our booth.
Yujan: Here are also some of our respected clients that you probably recognize at RSNA. We've had the privilege of helping them, so I wanted to give them a shout-out.
Designing for FDA Clearance 🔗
Yujan: Unfortunately, I only have 20 minutes for this conversation, and I can't possibly tell you everything we know in 20 minutes. What we're about to share with you is an average of lots of projects we've been through. It'll probably get you 80 percent of the way there; 80 percent of these topics will apply to your device. There's always that last mile problem of the last 20 percent that you should get some advice for. These are also highly opinionated. In no way is this an endorsement from FDA.
Yujan: I want you to take away from this conversation rules of thumb, nuggets of wisdom, 80/20 trade-offs, guiding principles, and the way that we have found success at Analytics doing this process. Please don't take this and plug it into ChatGPT. We've seen that happen; we had to clean that up. That's not going to work, unfortunately.
Statistical Power vs. Generalizability 🔗
Yujan: The golden road of medical device development might sound cliché, but I think it's important. You should design for others as if you design for yourself, and after that, find the least burdensome pathway to market.
Yujan: The first question that we get a lot is: Is being statistically powered enough to prove generalizability enough to get FDA cleared? The answer is no. Statistical power on its own does not prove that your device will generalize because it does not account for biases within the data—things like age, race, different types of devices. What FDA is looking for is reassurance that your device truly works, trained on your training data, tested on your test data, and will generalize when it gets into the post-market.
Yujan: Unfortunately, sample size, although that is one facet of what you need to prove, it's not the whole story.
How Much Data Is Enough? 🔗
Yujan: So then, how much data is enough? This is a very age-old question. Our rule of thumb on this—for non-CADx, so non-computer-aided diagnostic devices—200 subjects seem to be the sweet spot. If you need to do manual annotations, you need three readers. Make sure they're U.S. board-certified readers. Alternatively, you can have alternative ground truth sources like biopsy results, head-to-head predicate results. That can also be an option for you.
Yujan: My recommendation for how you should allocate your budget for testing data: Use 80 percent of your budget for the first big data pool. That should capture a lot of variability in your target data, but it's not going to capture all of it. That's what the remaining 20 percent is for—I like to fill in any gaps in that. So if there's any demographic gaps, if there's any gaps that perhaps are missing in the type of scanner manufacturer, or magnet strength, or whatever, that last 20 percent of your budget could be used to fill in those gaps.
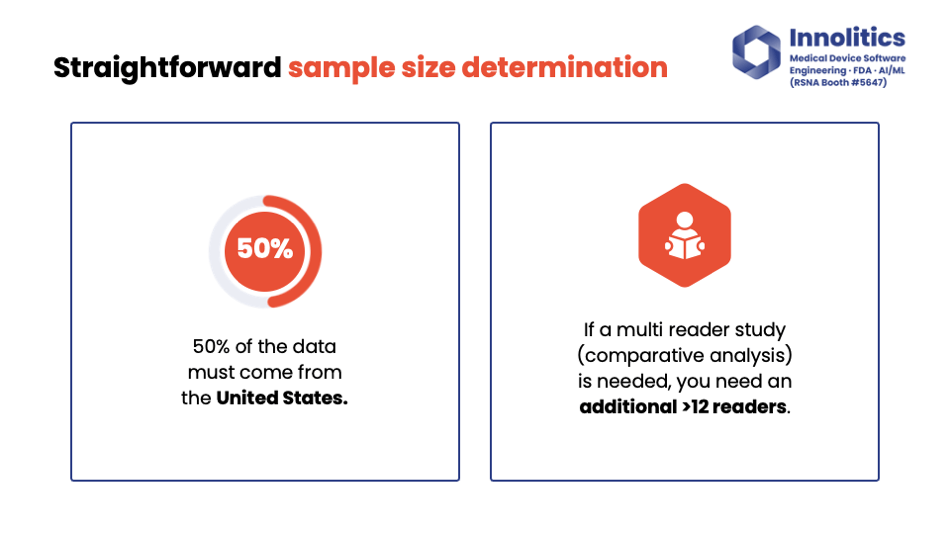
Yujan: The idea here is to identify potential sources of variability that could impact the actual performance of your device—things like imaging programs, acquisition systems, BMI if you're doing whole-body modification, for example. Make sure 50 percent of your data is from the U.S., and also, if you need to do a multi-reader study, make sure you're using at least 12 readers. That's been our kind of best practice.
Yujan: For your training data set, these principles are less required. Your training data set does not necessarily need all these. But for your test data set, that needs to be locked down pretty well.
Handling Ongoing Changes 🔗
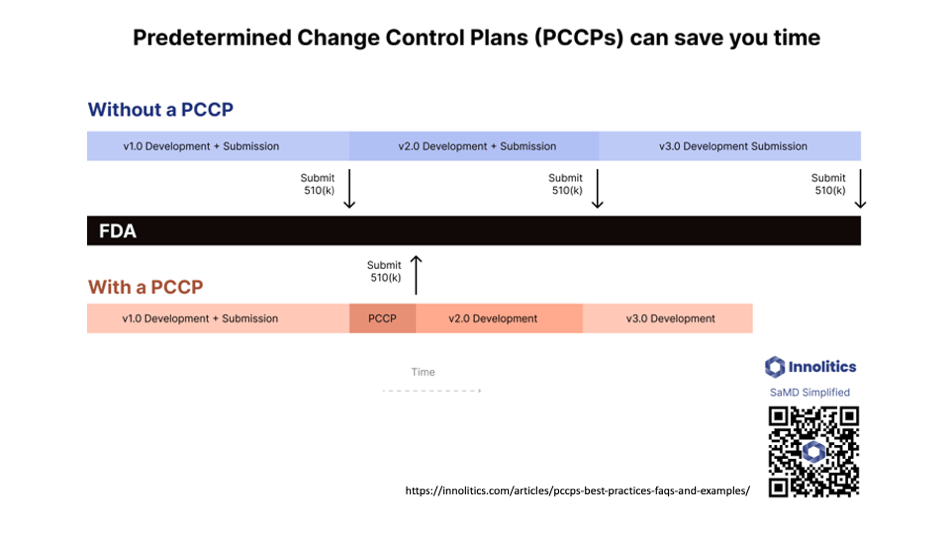
Yujan: Next question: To file or not to file? That is an excellent question. How to handle ongoing changes. There's draft guidance on this from FDA—or not draft; it's guidance from FDA—but it hinges on your interpretation of the word "significant." Is it significant enough? Is the change significant enough to require retraining your algorithm? In our interpretation, that does not constitute a significant enough change to require a new 510(k) application.
Yujan: If you want to play it safe, there are other avenues, like a Special 510(k) and also a PCCP, which is a Predetermined Change Control Plan. But if you're adding something new to your device, like a new disease condition or a new type of imaging modality, those probably are considered significant changes by the wording and would require a new Traditional 510(k), unless you've already done a Predetermined Change Control Plan with FDA where you pre-broker the deal. It's kind of like a pre-cert program for changes that you know that you're going to do, so you can do that in your pre-market submission.
Yujan: We have an excellent article on our website. If you'd like that, please go to our website and review. Or again, go to our booth for the post-show questions and answers.
Setting Performance Targets 🔗
Yujan: Performance targets are also a huge source of concern. Our rule of thumb again: If you're doing diagnosis or detection, shoot for AUC greater than 0.9. If you're doing a segmentation target, shoot for a DICE score greater than 0.8. For any sort of continuous thing, you can get these values; shoot for 5 percent of that value, 95 percent of it in agreement.
Yujan: However, you still need to justify to FDA how you got these values in the first place. In our opinion, there's three tiers of evidence that you can use to justify these values.
Yujan: The first and most preferred tier is when you can get your predicate device and see what they did. If they published performance targets, that's usually a very strong source of evidence to justify why you chose the targets you did.
Yujan: Failing that, you can also check the academic literature to see if there's any papers published on similar algorithms that have been done, and you can use that as further evidence to justify that your targets are meaningful clinically.
Yujan: And finally, if you have a particularly new quantitative imaging value that doesn't have a precedent, you could just measure your device on a pilot set, like a tuning set, and then you can set your targets to give yourself a little bit of a mirror mark. But that's the least preferred method.
Yujan: Also, outline the impact of false positives and negatives. So if there's a false positive—if you claim that there is a lesion and there isn't—what's the outcome of that? And that could be different based on what the clinical output is. What FDA is really looking for is the risk that is associated with your device.
Cross-Functional Teams and Trade-Offs 🔗
Yujan: At Analytics, we do R&D and also D&D on these machine learning algorithms. So our process—we don't transfer the algorithm out of the R&D step until we know with some degree of confidence on the tuning set that we're going to meet those aforementioned targets. And we will continue iterating on R&D until we do meet those targets.
Yujan: Now, unfortunately, not all devices have an unlimited budget or unlimited time frame. At some point, you'll have to get to a point where there's no more budget for another R&D round. In this case, you'll have to make some trade-offs. Trade-offs might not be as bad as you think. For example, a bounding box to outline the overall location of a tumor could be just as good from a sales and marketing and also clinical standpoint, but it could be ten times easier from an engineering standpoint. So you can make these little micro trade-offs as you go to find that sweet spot to creating something that the market wants at a reasonable price point.
Yujan: One other common thing that happens: there could be a subgroup where your algorithm performs very poorly, but that subgroup is something that's not really clinically meaningful. That subgroup is something that's not meaningful from a business perspective. So you can add in your labeling that your device doesn't support it or it supports it with less accuracy and give ground there where you can then save ground on some other topics that's needed.
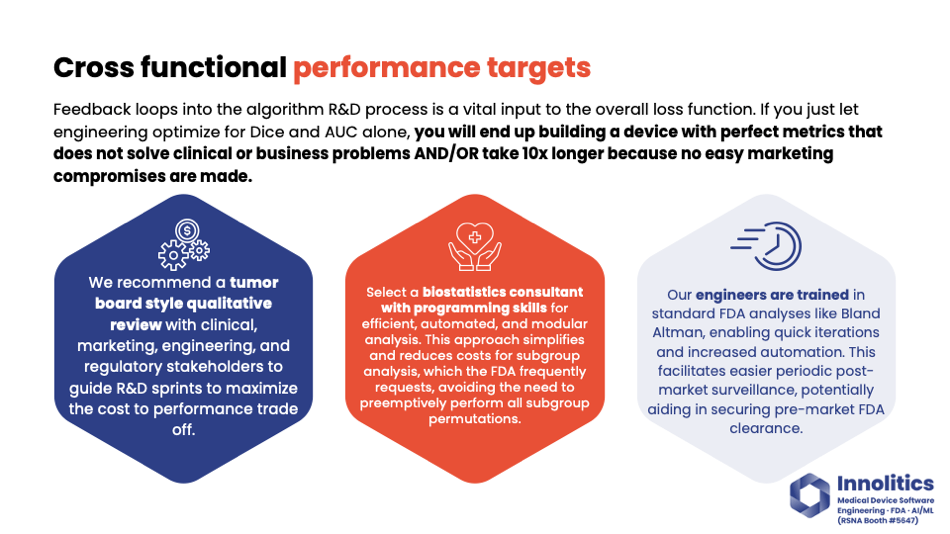
Yujan: It's important—kind of going back to that same point—it's important to have a cross-functional team. At Analytics, our process is to have, at the end of every R&D sprint, almost like a tumor review board. We go over qualitatively how the device is performing, and we put that in front of clinicians, we put that in front of marketing, regulatory stakeholders, because these folks know what the market wants, what will sell, what's going to be easy to make, and regulatory will know what's going to blow up the site design. Those viewpoints put together give you a much better loss function than just a DICE or an AUC alone.
Yujan: There's a lot of nuances that can't be captured in a DICE score. For example, if it's important to you to avoid blood vessels, a DICE score of 0.9 might actually perform worse than a DICE score of 0.8 in that case. You won't know unless you look at it.
Yujan: What we've seen, and we've designed our process to avoid this, is sometimes engineering will be in its own silo. They'll be chasing a DICE score, but in reality, you'll end up making a product that no one wants to buy, and you'll end up taking ten times as long and costing ten times as much.
Yujan: Also, biostatistics—we prefer to have biostatisticians with some amount of programming experience so that those can be fully automated, because you'll be doing a lot of subgroup analysis by this process. When you get your feedback from FDA, almost always there's a subgroup that you haven't done that you'll need to do. So having some quick mechanism to repeat analysis in a subgroup will save you a lot of headaches.
Importance of Pre-Submissions 🔗
Yujan: So, do I need a pre-sub? Yes. When's a good time for a pre-sub? We like to do it right after the algorithm prototype is done, so we have a good idea about the overall performance of the algorithm and we know how to design the study to go in that pre-sub.
Yujan: A consultant that has some software development experience is our preference because that consultant should be able to answer a lot of questions for us that you don't have to go to FDA for—things like cybersecurity and software validation. These are questions that don't need to go to FDA for ASAP.
Yujan: You should ask the high-risk stuff only, which usually for us is going to be the study design. The study design, to execute the study, could be a costly mistake to get wrong. These studies cost on the order of $150,000 to a million plus. So that's a high priority in order to ask FDA.
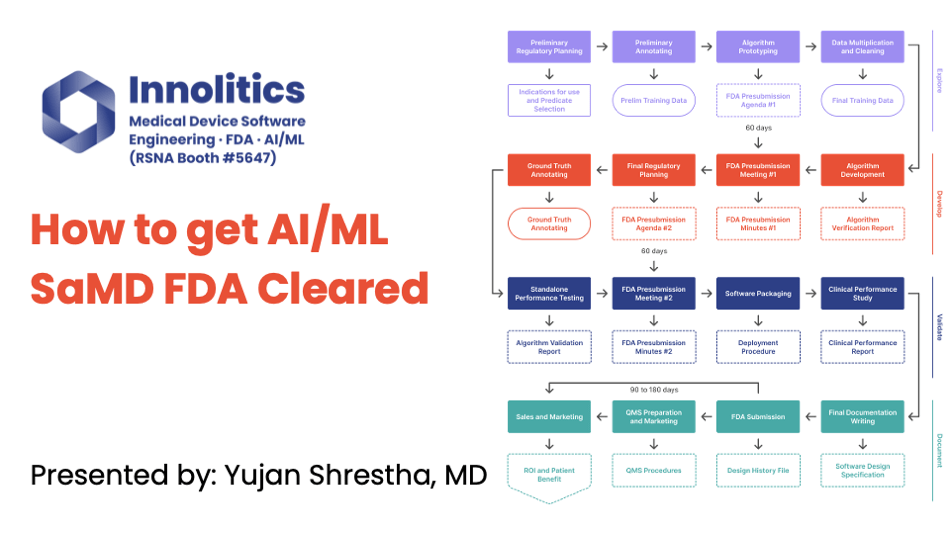
Yujan: It's important to communicate your algorithm design to FDA. FDA is very technical. They want to know how the algorithm works; they want to know all the steps involved, and they want to know the failure modes. So let's make their life easier by breaking out into different categories. Here's a block diagram key that we've developed. You can also look at more of this on our website.
Yujan: We like to split out the non-machine learning components from the machine learning components. This helps to fully communicate to FDA where the high-risk areas are, and they'll focus on the machine learning components whenever they look at this. They need to know what other components are there to make sure there's nothing hiding in the weeds.
Yujan: If you give FDA a lot of uncertainty about how your algorithm works, you're going to get a lot of uncertainty back. Or worst-case scenario, which we've seen a lot, is if there's a lot of uncertainty in FDA, they're going to give you the most conservative answer. And that conservative answer usually ends up costing you a lot more.
Yujan: Again here, I definitely like to map out the inputs and the outputs—this kind of diagram here. This allows FDA to see quickly what's machine learning, what's more traditional image processing. We have some more resources on our website if you want to see that.
Yujan: If you need help for a pre-sub, please stop by our booth. We'd be really happy to help you out. You only get one hour with FDA, so every second counts. And again, prioritize that study design.
Understanding FDA's Risk Perspective 🔗
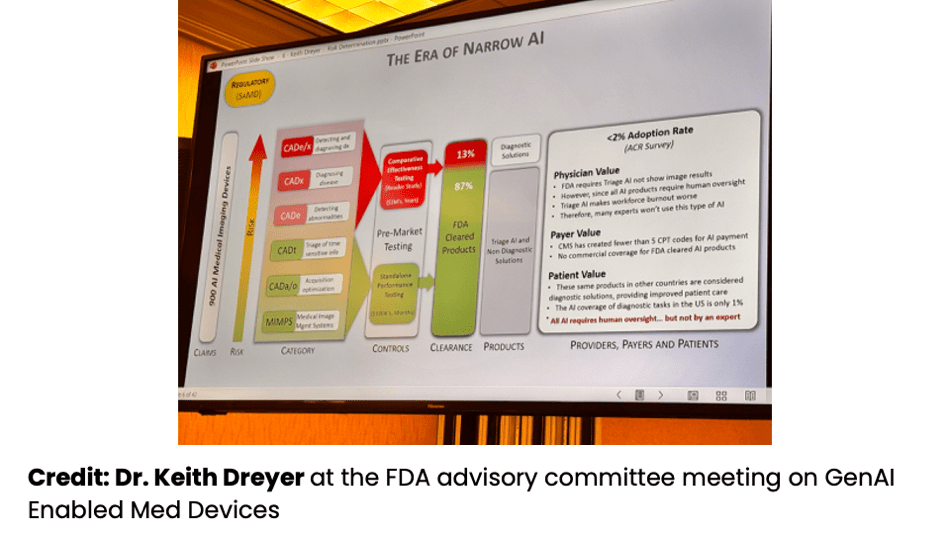
Yujan: I really like this diagram from Dr. Keith Dreyer back at the FDA Advisory Committee meeting a couple weeks ago. It clearly shows the level of risk involved in different types of AI devices, different types of CADs over there. You can see a clear demarcation of what's in red and what's in green. The red requires a multi-reader study, which—that's the ones that cost lots more money than just a standalone performance test.
Yujan: I'd like to break down what risk means to FDA in this case. I like risk; it's kind of like how much confirmation bias you are injecting into the practice of medicine, and there's two vectors to this. There's location bias—you're pointing a clinician to a specific spot—and there's diagnostic or clinical bias, where you're telling them or suggesting to them what to do next or what the diagnosis is. Those are two different axes, and you have a magnitude between those.
Yujan: Quantitative imaging devices, like product code LZQIH, have the lowest magnitude, where they only automate a process; they don't introduce that much confirmation bias into the practice of medicine.
Yujan: A computer-aided detection device, on the other hand, introduces a large amount of confirmation bias, but it's usually in the location axis, where you're pointing the clinician to a specific spot. You're focusing their attention there, so they might miss other spots. Or if you tell them nothing's there, they might not really go into more detail. That's the risk that FDA is looking for—is the confirmation bias that you have injected beneficial in the practice of medicine or not?
Yujan: On the other hand, computer-aided diagnosis—that's also injecting a large amount of confirmation bias, but it's going in the other direction. It's going in the diagnosis or in managing the clinical condition direction, and these two directions are not parallel. So you can have a device that does both. For example, if you have a device that will identify lung nodules, that's a CADx. If you have a device that does lung nodules and tells you if it's also cancer or not, that's a CADx and a CADi.
Yujan: Finally, there's computer-aided triage. This is the most restrictive of these here, but it could offer an easier path to market because the study design is a little bit simpler. But you can't inject confirmation bias in any of those two axes. You can't inject it on where the finding is; you can't inject it on what to do next or what the diagnosis is. You can only prioritize what images to review. So that may or may not work for your business use case.
Study Design for CAD Devices 🔗
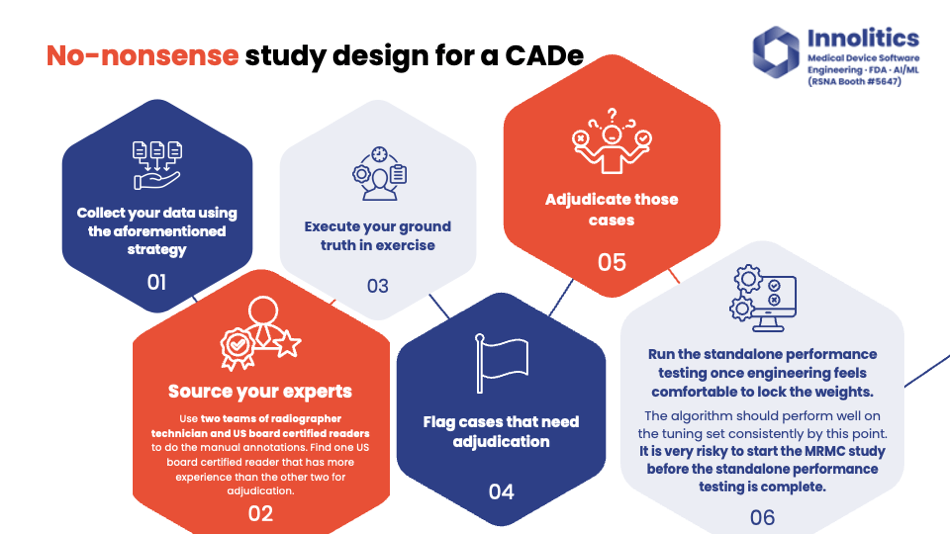
Yujan: For CADx design, here's an example of how a study is going to be done. First, you collect your data using the strategy we talked about. We like to use two teams of two experts to do the annotations. There's going to be some sort of technician that does pre-reads, and then you have a U.S. board-certified clinician that then does the QA step and does any editing.
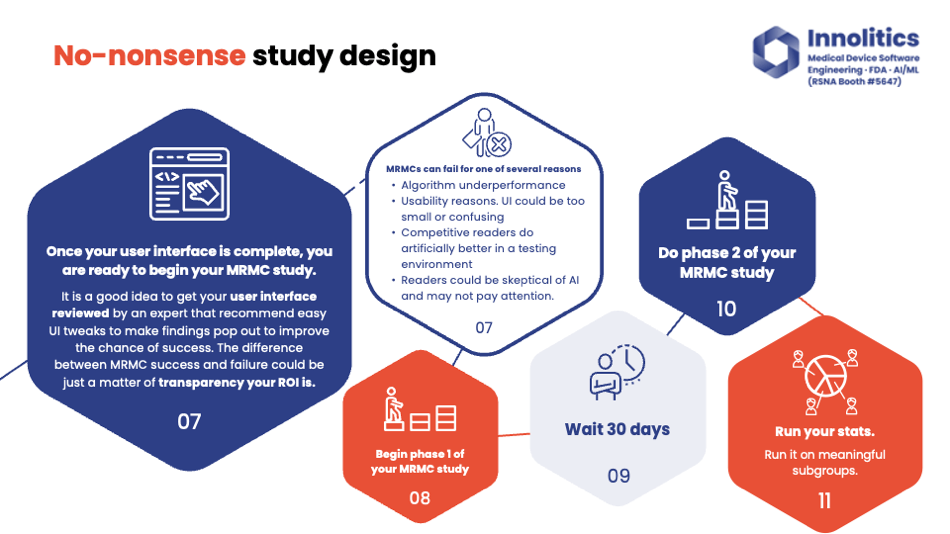
Yujan: Do your ground truth and also find any areas where the two teams of two disagree and adjudicate those ground truths. Importantly, only run the standalone performance study when R&D is completed. So when the engineering team feels satisfied with the current state of the progress, and they're ready to lock down the weights—they've run it on the tuning data and they're passing the more metrics there—then run the standalone performance study. Only then after that, run the multi-reader study. Once you've passed the standalone performance test, it could be an expensive mistake to run the multi-reader study too early.
Yujan: So multi-reader study could fail for one of several reasons. The first reason: your algorithm just might not perform that well. If you did your standalone performance testing first, you would be able to find that out before doing this multi-reader study. So in this case, standalone performance testing works well or doesn't work well, and your multi-reader study will either.
Yujan: Also, there could be subtle tweaks to your UI that could really improve this multi-reader study. Perhaps your tumors are just too transparent; it's really difficult to see, or small lesions are really difficult to see, so you might want to put an arrow there instead of just a dot. Things like that—you would see a high standalone performance test, then a low multi-reader study output.
Yujan: Also, some readers could just be skeptical of the AI and may not really want more confirmation bias or want additional information. In that case, too, your standalone performance test would be high and multi-reader study would be low. There's typically a 30-day washout for the standalone performance test, which you can see why this one is much more expensive than the previous slide that we did.
Final Thoughts and Generative AI 🔗
Yujan: Some cybersecurity—you can learn more about that on our website. Basically, the gist of it is, be sure to involve your software engineering team in this. Our engineers are typically—like, if we can't come up with low-hanging fruit to properly solve cybersecurity issues, there's something deeper, or I feel like we're going a little bit too deep into that topic, and you should be able to justify to FDA when you don't need to do that.
Yujan: Human factors—if you don't have any critical use cases, you don't need it. User needs validation survey—simple if it's a low-risk device. Automated tests—we should use those. Sorry, I'm running out of time, so I'm going to go through some of these here.
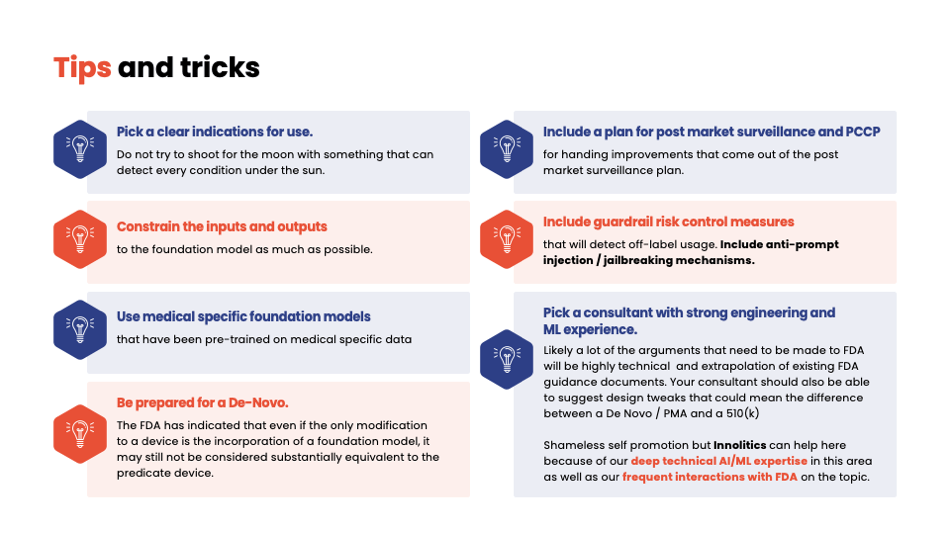
Yujan: So some things about GenAI and foundation models. Currently, nothing GenAI has been cleared by FDA, and just because your device does the same thing that your predicate that doesn't use GenAI does doesn't mean it's going to be substantially equivalent. Jury's still out on that.
Yujan: Here's some tips and tricks for GenAI stuff. Clarification—don't do everything under the sun. Constrain your inputs and outputs. Always keep it so that the user can't just use you as a ChatGPT, where you have not cleared indications that are being used. Include things like anti-prompt injection and anti-jailbreaking attacks. And also, just a little plug, pick a consultant with strong engineering experience like us.
Yujan: So yeah, thanks for your time. Hope you learned something. And again, if you did, please continue the conversation right over there at our booth. Thanks a lot.
