We Mapped the FDA Pathway for an AI/ML Ophthalmology Division Launch
Large Industrial Company
FDA Regulatory ConsultingAI/ML
The Problem
A large industrial company is looking to revolutionize the ophthalmoscopic OCT market. They're looking for an expert to help them strategize on the pathway to least resistance to an FDA clear product and provide software development coaching to create a new software division that is capable of producing FDA medical device grade software and is able to do so independently of outside consultants. They were looking for consultants that can help them write their SOPs, provide guidance on AI/ML strategy, and submit an FDA pre-submission.
The Outcome
We understand that regulatory strategy and business strategy are interlinked. We were able to provide insights on the regulatory and engineering part of the equation, allowing the client to balance business needs such as the amount and timing of funding raises, the planning of design and development, and other strategic partnerships and licensing deals necessary to bring the device to market. We identified a phased regulatory strategy approach of first bringing in lower risk indications and slowly adding additional hardware and software elements with downstream FDA applications in order to maximize chance of success with the given funding and future planned funding rounds.
The Solution
The FDA pre-submission meeting deliverable contained the following:
A submitted presubmission request. FDA will take 60 to 75 days to review after submission.
Meeting Purpose
Device Description
For each algorithm component:
Algorithm description and verification strategy
Standalone Performance Testing Plan
Clinical Performance Testing Plan
Training Data Description
Demographics, sample size, number of Institutions, qualification of the annotators
Validation Data Description
Demographics, sample size, number of Institutions, qualification of the annotators
Proposed Predicate Device
Proposed Indications and Intended Use
Predicate Device Comparison
Summary of business goals
Key marketing claims
Draft intended use statement
Possible pathways to US market (e.g., 510(k), De Novo, PMA, etc.)
If a 510(k), possible predicates
Key regulatory risks (i.e., issues that may prevent getting the SaMD to market, and how likely they are to occur)
Strategy related to future submissions (although the focus of the strategy will be on the first submission)
A clinical study design protocol with the following:
We work with your engineering and clinical team to set reasonable performance targets
We choose the best performance metric to use (e.g., Dice, mean absolute error, surface Dice)
We decide the best ground truth annotation strategy.
The process used to produce the deliverable involved asking a series of questions. Here are some examples:
Understand Business Goals
What are you trying to accomplish by bringing the SaMD to market?
What markets do you want to sell it in and what is their relative importance?
What marketing claims do you need to be able to make about the device’s performance to accomplish these business goals?
How will the software be used (e.g., within the clinical workflow or directly by patients)?
Draft a rough intended use statement for the device.
Understanding Software and Clinical Workflow
What work has been done on the software?
What do you hope to implement in the next year or two?
What functionality is the highest risk?
Create Technical Description of Algorithm to Maximize FDA Feedback
Step 1: Algorithm Runtime Description
Step 2. Datasets Description
Step 3: Non-ML Verification Testing
Step 4: Historical Annotation Description
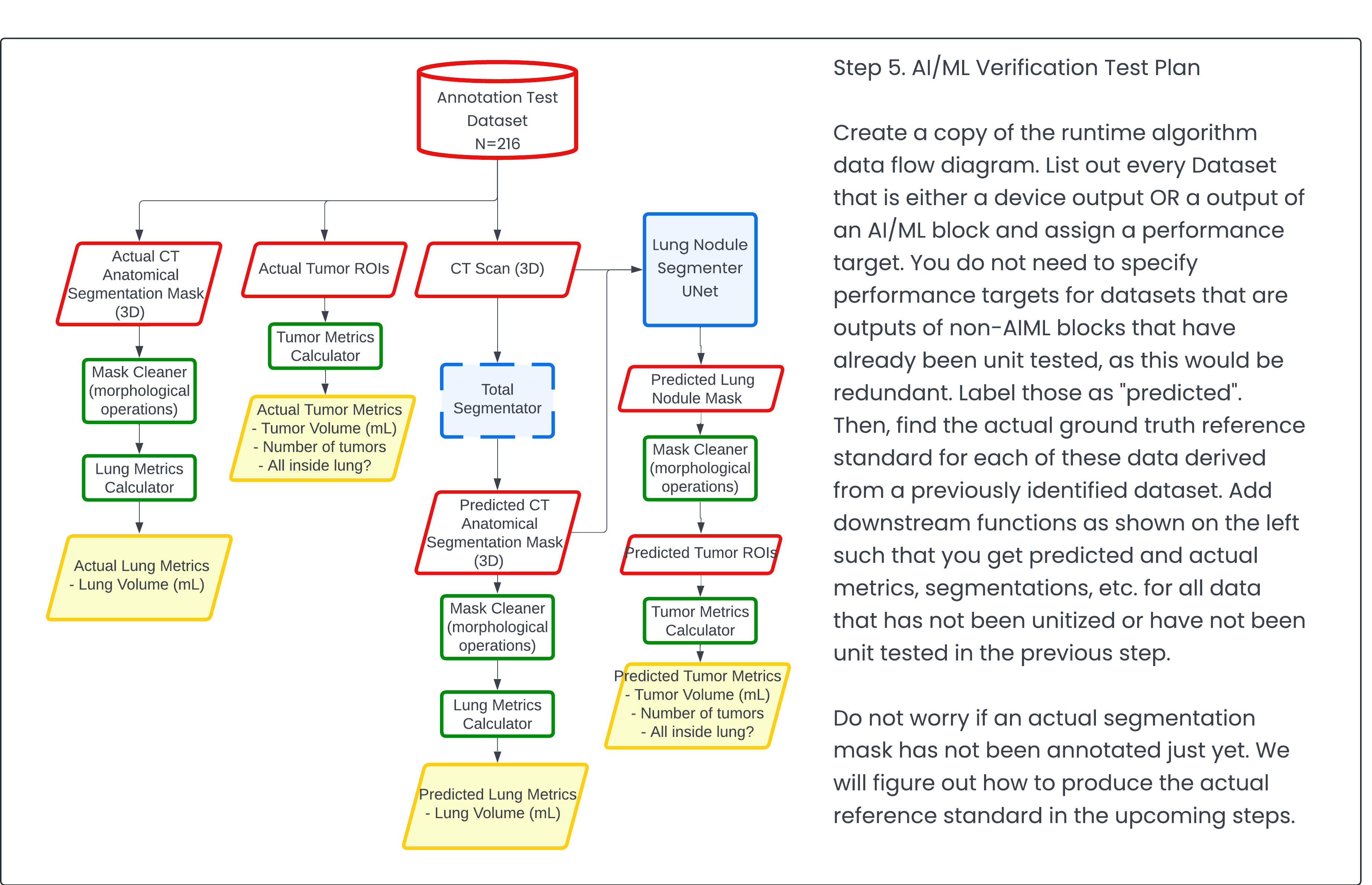
Step 5: AI/ML Verification Test Plan
Step 6: New Annotation Procedure Description
Step 7: Performance Metrics
Develop Regulatory Strategy
Determine the product code for the device
Identify applicable FDA guidance and Standards
Determine available market pathways (e.g., 510(k), De Novo, PMA)
If a 510(k), identify possible predicate devices.
Will the SaMD need clinical performance testing, and roughly what form this would take?
Identify key regulatory risks and ensure they’re inline with the business goals.
Plan pre-subs meetings with the FDA and how we’ll mitigate regulatory risks
If the device is a series of modules will each module have it’s own IU statement? Will there be an overall software ‘Platform’ which serves as the user interface and which may perform some functions (i.e., image viewer, report generation)?
If there will be multiple submissions (this is common for SaMD), what order should we complete them in?
If the device involves AI/ML, do we want to include a PCCP in the first submission?
Develop a Clinical Validation Strategy
Determine if the product is a measurement only, CADe, CADt, or CADx device.
Explore the data you already have available to ascertain if it can be reused for the study.
Work with you to tweak your device’s indications for use and marketing claims to make the clinical validation study significantly easier and faster without sacrificing too much marketing value.
Conduct a statistical power study to justify the anticipated sample size.
Design the standalone performance study design
Design the clinical performance study design if necessary
Provide a proposal for the cost necessary to conduct the study(ies)