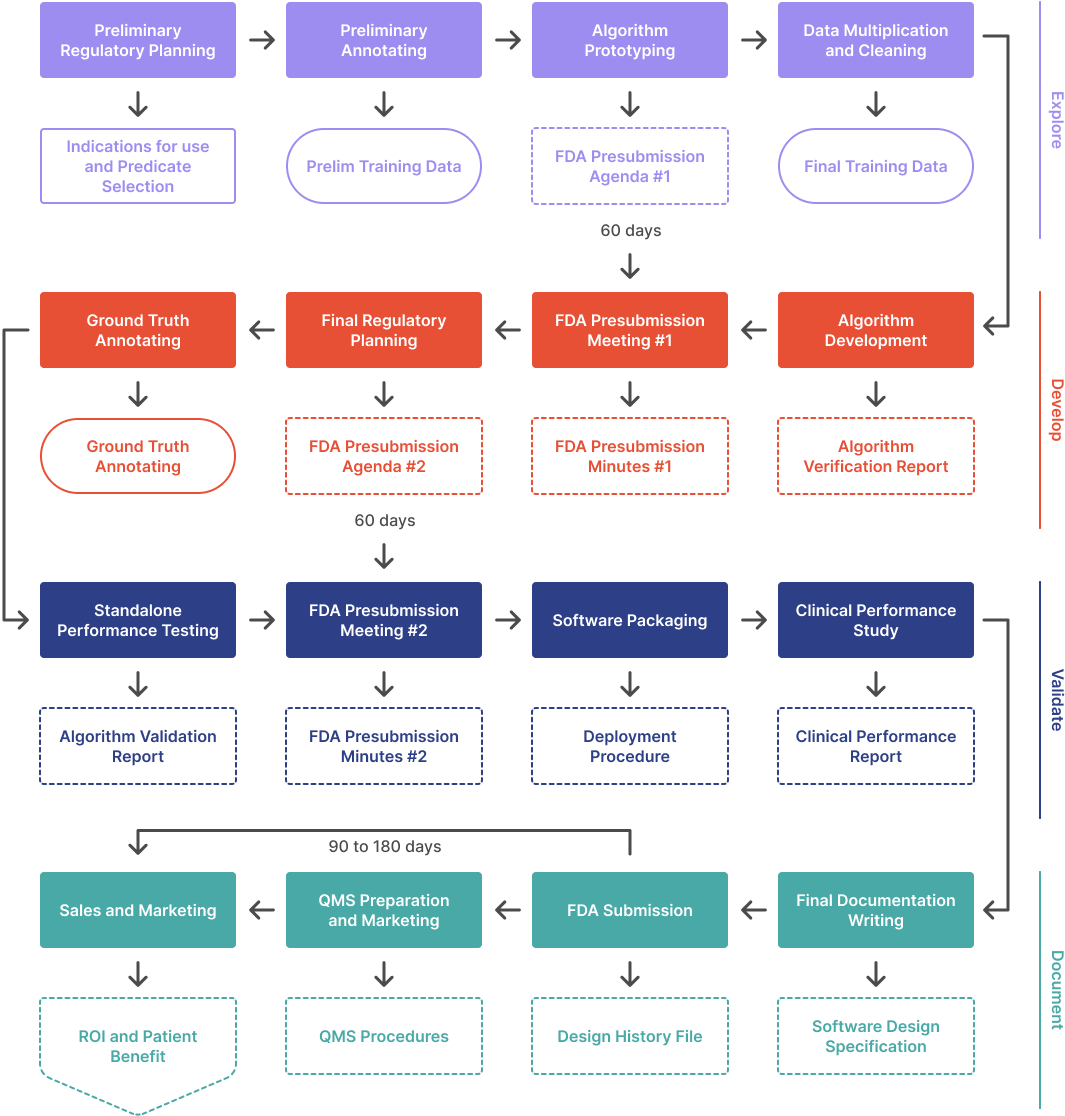
Overall Process 🔗
Is this article for me? 🔗
This article outlines the process to develop an ML/AI algorithm from scratch and get it FDA cleared. It covers the four phases of the process: Explore, Develop, Validate, and Document. It also discusses the costs, time, and data requirements involved in the process. Additionally, it provides advice on regulatory strategy, data annotation, and algorithm prototyping.
This article is for you if:
- You have an idea for an algorithm and want to explore ways to gain the necessary FDA clearance to bring it to market. This article describes how to write an algorithm from scratch, but if your algorithm is already built, you can skip the prototyping, development, and first-presubmission steps.
- You are creating a Quality Management System (QMS) procedure to comprehensively and clearly explain your company’s process for developing AI/ML medical devices.
- You have recently been acquired, and your parent company has asked you to improve your AI/ML design and development process to ensure a high level of quality and safety.
- You are in a startup accelerator program and need some help with the process of developing and bringing a medical device to market involving AI/ML.
In this article series, we outline the process to develop an ML/AI algorithm from scratch and get it FDA cleared. This article covers the first phase in a four phase process:
- Explore. Figure out what is possible and build a prototype. Meet with the FDA the first time.
- Develop. Set performance targets, implement, and iterate. Meet with the FDA again.
- Validate. Wrap up the design and test your product in a clinical setting.
- Document. Prepare and submit your 510(k).
The typical AI/ML Software as a Medical Device (SaMD) project takes about 12 months to complete. The main factors affecting the timeline are the complexity of the algorithm, how much you have already developed, and the quality of your existing documentation. Although the FDA’s target review goal for traditional 510(k)s is 90 days, expect the process to take 180 days when you factor in responding to FDA’s questions.
Note that these estimates exclude costs related to data acquisition, data annotation, or any necessary clinical performance study.
If you follow the process in this article, you can have an FDA cleared product on the market in as little as nine months—even sooner with a little guidance from us.
Step 1: Preliminary Regulatory Planning 🔗
A common mistake is to rush into implementation without first developing a regulatory strategy. Consulting a regulatory expert can save time and money. For example, it may not be obvious, but submitting an initial 510(k) with a simpler set of features followed by a 'Special' 510(k) with more complex features can be an easier path to market a device. This approach is often simpler than obtaining an initial 510(k) clearance for a device with advanced features. You may approach regulatory strategy in the same way they would write an SBIR grant. However, good FDA submissions require a more conservative, straightforward, and methodical approach. This approach can be in opposition to writing an innovative, exciting, and plausible SBIR grant.
I would encourage you to search for similar 510(k) submissions using our tool or the FDA database. Previous 510(k) submissions will give you an idea about what is possible with the current state of the art and reveal key insights on the clinical performance targets and study design. While the information that is publicly available on the 510(k) summary will not go into proprietary details, you can read between the lines to infer why the competitor made the decisions they did. This could save you from making the same design mistakes and give you a hint about the number of samples you should expect to annotate.
The deliverables of this activity should be the following:
- Indications of use / intended use statement / target patient population
- User needs
- Predicate device selection
- Rough clinical performance targets
Step 2: Preliminary Annotating 🔗
An AI/ML needs a significant amount of expensive clinical data but you do not need all of it to test the feasibility of the algorithm. Publicly available datasets such as the cancer imaging archive could be a good place to find unlabeled or preliminary annotated data to augment the dataset. Public datasets are useful to prove the concept but may not be viable to use for the 510(k) submission because the data provenance may not be specified or the licensing scheme may be unfavorable for a commercial product. I would also recommend staying away from competition data because the license usually prohibits commercial usage. Additionally, commercial data brokers exist that can procure the data for a fee.
How much data do you need to start out? There are several considerations. To use training data efficiently, there are some tricks you can do. In my experience, a segmentation problem needs less annotated data than a classification problem to achieve a comparable performance. It may be beneficial to convert a classification problem into one that has a segmentation intermediate. For example, if you are training an algorithm to detect contrast in a CT scan, one approach is to train a binary classifier on the entire CT volume. Alternatively, you can segment the aorta and kidneys and measure the average HU value. If the average HU value is higher than a threshold, there is likely contrast in the image. I estimate that the former approach will take 20x more images than the latter to achieve the same performance. Although it takes longer to annotate each image with a segmentation instead of just a yes/no checkbox per image, you will need to annotate fewer images. This results in a more transparent algorithm, which the FDA prefers. Therefore, I think the tradeoff is worth it.
Don't get too caught up in sample size calculations and estimates just yet! It's more important to get the project started than to get everything perfect right away. Perfection can be the enemy of progress.
Step 3: Algorithm Prototyping 🔗
Once you have your preliminary data, now is the time to start developing a proof of concept. This has several advantages:
- It is difficult to estimate the expected accuracy of the algorithm without building a prototype first. Although algorithm accuracy does improve with more data, 80% of the performance is often reached with just 20% of the data.
- It is difficult to estimate the project timeline and budget without building part of the product first. Having an informed budget and timeline will be important if you need to find an investor or write a grant.
- It is difficult to estimate the number of images required for training without building a prototype first. if you find the prototype already has reached clinically useful performance then congratulations! The performance will only get better from there. If the prototype is getting poor results, this gives you an opportunity to pivot before you have sunk more money into a potentially unsolvable problem, reevaluate the accuracy targets in mind, or clean up the input data.
- It is difficult to elucidate all the edge cases we may encounter without playing around with some data first. An algorithm approach that should work well on paper may not perform well with real world data. In other words, building a prototype helps convert “unknown unknowns” into “known unknowns” for the engineers and clinicians involved in the project. Engineers learn about what failure modes are the most clinically detrimental and clinicians learn what intended uses are most plausible.
The output of this step is the preliminary algorithm development plan, which can form the content of the first optional FDA presubmissions meeting.
- You can skip this presubmission meeting if you have identified a closely matching predicate and have a good idea of your clinical performance metrics. We still recommend you have a presubmission meeting a bit later in the process before you start your clinical performance study.
- A presubmission (presub) meeting is a free meeting with FDA to introduce your AI/ML device and to obtain specific feedback early on in development. Presubs are particularly important to:
- Confirm your proposed predicate device. FDA will provide feedback related to the viability of the chosen predicate and to its suitability to be used to determine if your device is ‘substantially equivalent’ the the predicate device.
- Confirm the regulatory pathway. If there are no substantially equivalent devices on the market, FDA could suggest the De Novo pathway may be appropriate route to market.
- Confirm the preliminary design for your clinical performance study. The clinical performance study could be more expensive than the software development effort.
The meeting must be scheduled, and typically occurs between 60-75 days from the date the request is submitted. The meeting can be in person (rare) or virtual (typical). You may also elect to have FDA provide a detailed written response to your questions in lieu of a meeting.
- Overall algorithm design. Will there be a neural network? What other image processing techniques are used? What are the algorithm components and how can their performance be individually measured? Example: Algorithm shall consist of a contrast classifier, Unet based segmentation neural network, and postprocessing steps using morphological operations. The contrast classifier shall be evaluated with a binary cross-entropy score. The Unet shall be evaluated with a Dice coefficient. The morphological operations need not have performance targets and instead is verified with unit tests using synthetic data.
- True positive definition. What criteria will you use to consider an algorithm output good enough to be clinically useful? Example: lesion centroid must be within 5mm of the ground truth.
- Preliminary performance metrics. What marketing claims do you think are possible and clinically useful? Example: device should achieve a sensitivity greater than 95% and false positives per image of less than 1.
- Proposed clinical performance evaluation study design. How many clinicians are required to annotate the ground truth? What will the study design look like? What is the clinical performance target and how will you prove your device meets the target? How are you accounting for common biases? Example: Three radiologists shall independently annotate the ground truth used for the clinical performance stud. A neuroradiology specialist will adjudicate any annotation that does not achieve consensus. 21 different radiologists will be invited to review 400 unlabeled images for lung nodules. After a washout period of 30 days, 21 radiologists will review the same 400 images but this time will have the algorithm outputs showed concurrently.
Step 4: Data Multiplication and Cleaning 🔗
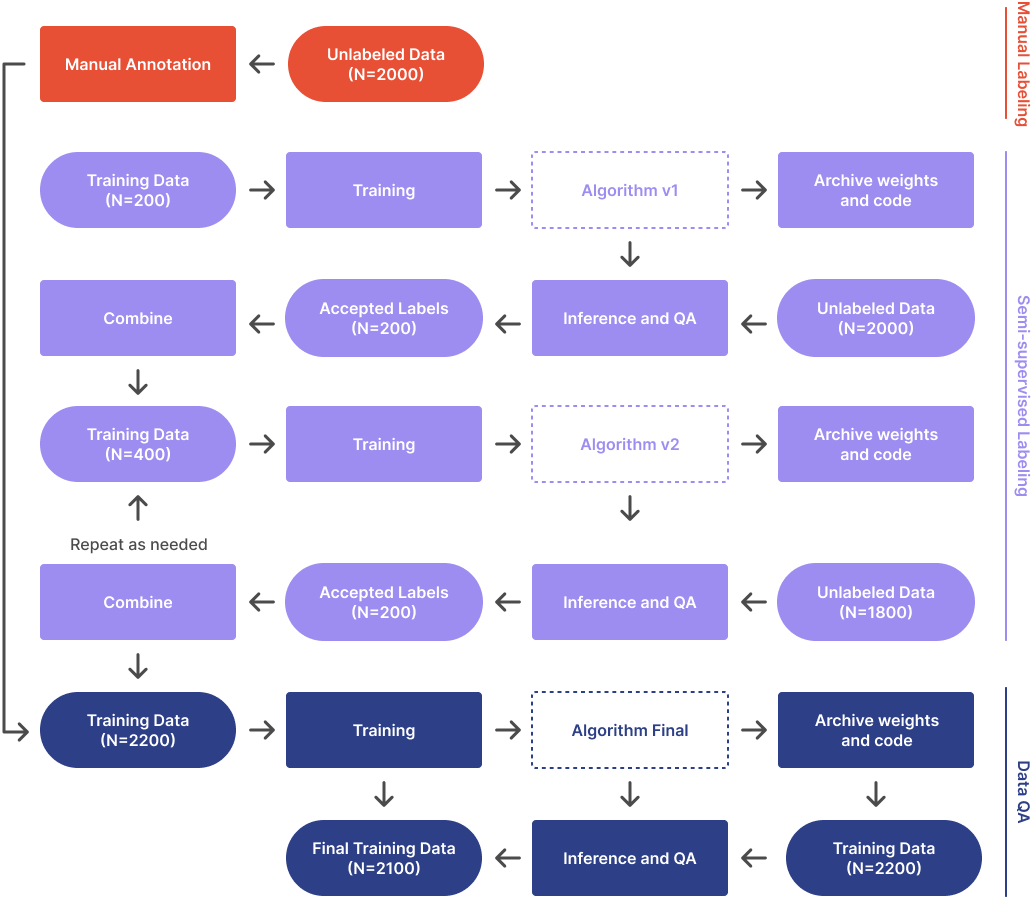
At this phase you should have have the following:
- Expect that the final version of the algorithm, with more training data, will produce clinically useful results. If the results are unsatisfactory, consider changing the algorithm or adjusting the intended use. For example, if the Dice scores are poor but the centroid distance metrics are acceptable, switch the intended use to display only the center of the tumor instead of the outline. It is better to show less information than noisy information. Reevaluate whether the chosen predicate device is still viable.
- A prototype you can show investors or other stakeholders in case you need additional funding to procure and annotate additional training data.
Once you have procured about 10 times the preliminary training dataset, you must annotate them. There are several ways you can achieve this. One option is to continue to annotate manually. An alternative is to run the preliminary algorithm on the unlabelled dataset and manually QA the results. Then retrain the algorithm with the larger training set. Repeat the cycle a couple of times until all the data is labeled.
Once all the training data has been annotated, it is a good idea to QA the data to detect errors. I recommend having a clinician review the annotations. Additionally, running inference on the training dataset and flagging any annotations that show significant errors is beneficial. It may seem like poor practice to run inference on training data, and it is if we are using the results to make performance claims. However, this run is only to detect outliers in the training data that could indicate manual annotation errors—we are not making any performance claims.
Step 5: Algorithm Development 🔗
At this step you should have the following:
- A full set of training data that has been manually checked for errors
- A good idea of your clinical performance targets and key marketing claims
- An algorithm prototype you can continue to iterate on
- An optional presubmission request pending with the FDA.
In this step, explore frameworks, neural network architectures, tweak parameters, and get the most performance out of your algorithm that your budget allows. There is no upper limit for the budget, but there is a point of diminishing returns. Keep the end goal in mind: create something clinically useful and marketable. Otherwise, it's easy to continue iterating for academic reasons—a trap I've fallen into. We discuss these potential traps in a prior article. This is the most enjoyable part of the ML development lifecycle, but it also has the most ambiguous endpoint. Here are some pointers:
- Stop once you have reached your clinical performance target. Anything beyond this is may not be the best appropriation of resources. This is an appropriate time to define the ‘minimally viable product’ (MVP). Defining the MVP includes drafting the marketing claims which are seem as ‘must haves’ in order to sell the product and compete for market share.
- Is there a population of cases that could be problematic, such as very large lung nodules, if removed, and would mean you meet your clinical performance target? If so, would the clinical utility of the device significantly diminish if this problematic population is excluded from the device's intended use? Consider how much it may cost to increase performance for this population to evaluate if the cost-benefit ratio makes sense to continue iterating. Unfortunately, this is a research problem and it is often impossible to accurately estimate the unknown. However, a literature search and deeper predicate device search can give you some clues.
- Can your clinical performance targets be adjusted to meet the threshold of clinical utility and marketability? For example, you may be detecting the presence of lung nodules but not getting the outline correct. In this case, your Dice score will be low, but your detection sensitivity may be quite good. This would mean you lose the ability to draw an outline in the UI, but you could draw an arrow instead. This could still be clinically useful enough to take the compromise and move on. It is important for engineers to get a rough idea about clinical utility, so they can consider these tradeoffs without needing to involve a clinical expert.
- Rapid iteration with clinical stakeholders is essential. To facilitate this, use tools that both clinicians and engineers can use, such as Excel spreadsheets or Notion databases. Every round of feedback presents an opportunity for knowledge diffusion between engineering and clinical teams, increasing the likelihood of "building the right thing" rather than simply "building it right".
Also note, the FDA will likely get back to you with a presubmission meeting date during this step.
Step 6: FDA Presubmission Meeting I 🔗
FDA sends responses to your questions before the meeting so you already know “what” they think prior to the meeting. It is important to carefully read and understand all of the information before the meeting. In my opinion, the best use of the 1 hour meeting is often to ask FDA ‘why’. For example, if FDA disagrees with your choice of a predicate device, it is extremely helpful to understand why. Perhaps they feel that the technology is too dissimilar that it wouldn’t be possible to effectively compare the devices. It may be possible to justify your choice and gain their agreement or they may provide examples of suitable devices.
In my opinion, the best way to utilize the first FDA meeting is to ask the FDA “why” so you can learn how to think like the FDA when they are not immediately available for inquiry. Make sure your team is taking notes because you are not allowed to record the call and are required to submit meeting minutes to the FDA for approval.
Step 7: Final Regulatory Planning 🔗
At this point of the process, you should be able to:
- Finalize the clinical study design and procedure in full detail, including any protocols, processes, and timelines.
- Finalize clinical performance targets
- Finalize algorithm design
- Finalize ground truth annotation procedure
Output:
- Agenda for your second FDA presubmission. This meeting will cover topics at a greater level of detail than the initial meeting which was held early in development. Questions for FDA at this phase will likely be related to:
- Confirmation for reader qualifications/experience
- Confirmation of statistical analysis
- Confirmation of bias mitigation. This often involves visualizations of your dataset including age, sex, acquisition device manufacturer, etc.
- Confirmation of key performance targets.
- Confirmation of clinical validation plan. Conducting a clinical study can be very costly, and the FDA may reject the output, leading to the downfall of many AI companies.
Step 8: Ground Truth Annotation 🔗
- The ground truth used for the clinical performance study should be of a higher standard than the data used for model development. This dataset will be used to demonstrate to yourself and the FDA that the device is meeting its intended purpose.
- Consider conducting a cost-benefit analysis of the annotations. If it takes 5 minutes to do a point annotation, but 20 minutes for a polygon, consider rewriting your acceptance criteria to focus on detection rate versus Dice score. Arguably, the former provides 95% of the clinical utility for 20% of the cost.
- Ideally, the ground truth should be kept separate from the engineering team to avoid accidental use in model development. However, guarding against deliberate data leakage—engineers deliberately using data from the ground truth for training—is too burdensome because engineers usually need to handle the ground truth to calculate performance metrics.
- The FDA likes to see that exclusion criteria are thoughtfully considered, and that images aren't thrown out for arbitrary reasons. There are a few reasons why images may be excluded from the ground truth dataset, such as image quality and presence of artifacts. To minimize bias, consider including images with varying levels of difficulty so that the device will be tested on all levels of complexity. Additionally, if possible, consider using an independent adjudicator to review any images with annotations that did not achieve consensus among the annotators.
Step 9: Standalone Performance Testing 🔗
- Run your algorithm on the annotated ground truth data and verify you are meeting your standalone performance metrics. You should have a high degree of confidence of model performance already with the dataset used for development (train, validate, test)
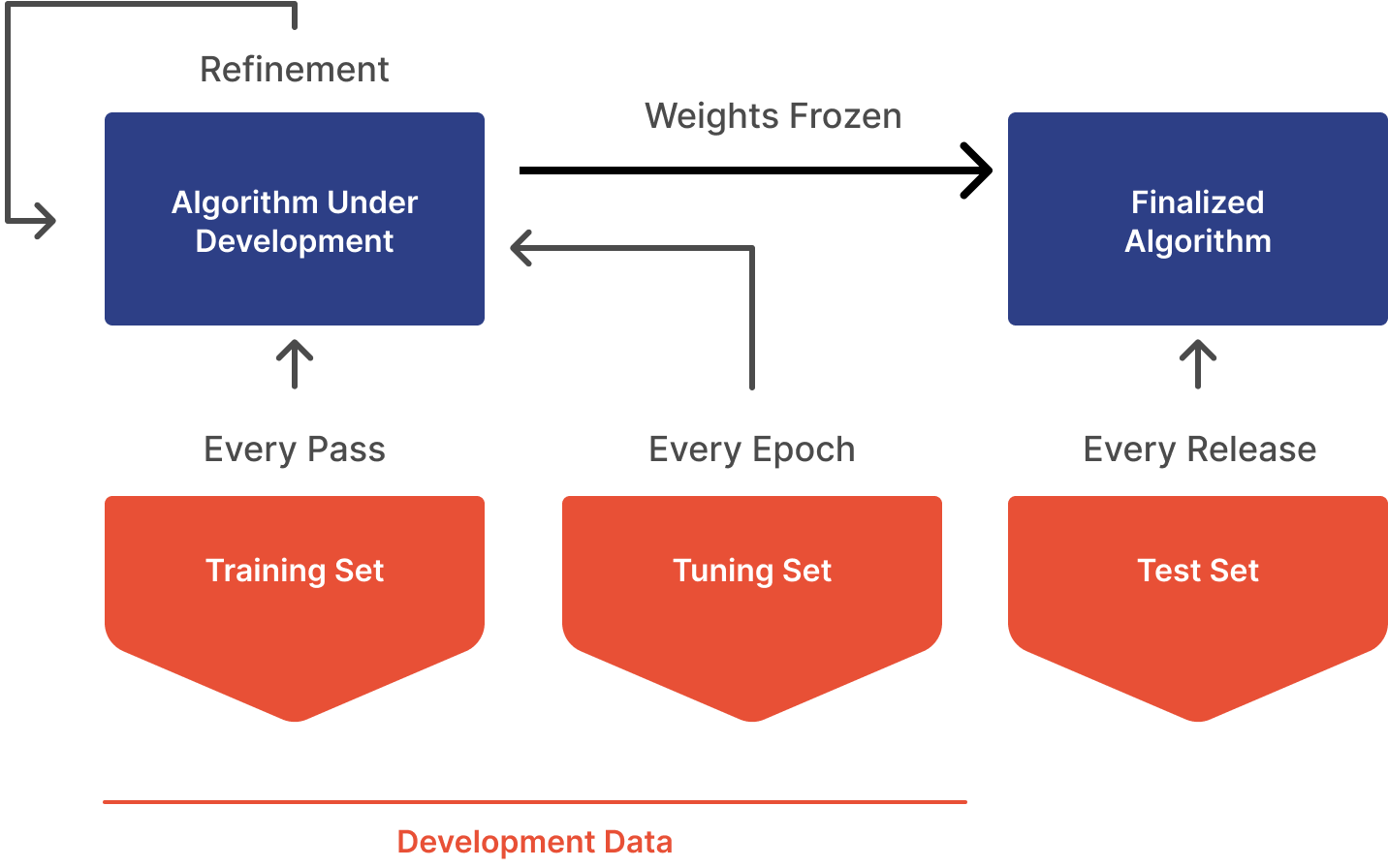
- Unfortunately, the terms "validation" have different meanings in engineering and regulatory contexts. In engineering, the validation set is the data partition used to detect overfitting to the training set. This set is usually tested against the model every epoch. On the other hand, the clinical performance set is used as part of the medical device "validation" process, which is done on the completed device after the model weights are frozen. This process is different from validation in the engineering sense.
Step 10: Presubmission Meeting II 🔗
By now you should have the results of standalone performance testing. You should let the FDA know if you had to change the standalone performance testing targets. The main goal of the second presubmission is to make sure the FDA:
- Agrees on the validity of your clinical performance study design. It is helpful to know that FDA has recently released a final guidance related to their expectations for clinical performance assessments for 510(k) submissions.
- Agrees the study proves your device is performing to its intended use, meeting the proposed indications for use and supports the key marketing claims.
The clinical performance study can be very expensive. You will likely need to pay many highly trained individuals to sit in front of a computer for hours on end and get them all together in a room to debate tough cases. Pizza helps but it is usually insufficient on its own.
Note that clinical trials are different from clinical performance studies. A clinical performance study has a lower burden of proof than a trial. Whereas a trail needs to prove the benefits of the device outweigh the risks, a performance study just needs to prove the medical device is performing as intended in a real world clinical setting.
FDA may have some concerns such as:
- concerns about biases in ground truth data collection. For example, your ground truth data collection strategy may be missing any attempt to include more than one institution, CT machine manufacturer, patient population, or other confounders.
- concerns about the performance study protocol. For example, you may have not included a washout period to account for recall bias.
Again, ask “why” so you can think like the FDA.
Step 11: Software Packaging 🔗
The software release package should include a list of all the software components and versions. It should also include a list of all the external libraries and their versions. This is important for maintaining compliance with the FDA's off the shelf software requirements. The deployment package should include detailed instructions for setting up the software and deploying it to production. The validation package should include a list of tests which should be run on the software and their expected results. The documentation package should include documentation for all the components of the software and their interactions.
It is best practice to package the software for release on a continuous basis, ideally nightly. Docker is the most common choice for this. Clinical performance studies should be conducted on a version that is as close to complete as possible. If software changes are made after the study is complete, it is necessary to justify why the changes do not affect the result. We strongly recommend that no software changes be made to the AI/ML components after the study is finished.
Step 12: Clinical Performance Study 🔗
Perform your clinical performance study. Make sure that the study design is in compliance with the FDA's final guidance related to their expectations for clinical performance assessments for 510(k) submissions. The study should prove that the device is performing to its intended use, meeting the proposed indications for use, and supporting the key marketing claims. It is important to consider any potential biases in the ground truth data collection, as well as any risks for recall bias when creating the study protocol. Additionally, software changes should not be made to the AI/ML components after the study is complete, and the software should be packaged for release on a continuous basis, ideally nightly.
Step 13: Documentation 🔗
We left out some key documentation exercises done earlier on in the process to keep the deluge of information at a manageable level. Risk analysis and requirements capture should be done during the development process but it is not uncommon to be done retrospectively—which can be a daunting task if you have not done it before. We can write your FDA documentation for you if you do not have the time to do so yourself.
We will cover the documentation aspects for creating 510(k)s for software devices in a future article “Content of a 510(k) for software devices”. In the meantime, check out FDA’s guidance document on the topic.
Step 14: FDA Submission 🔗
The submission step is the culmination of your development, testing and documentation efforts. We recommend that you have someone help with the creation of the submission as a successful submission is much more than a compilation of design and testing documents. A 510(k) is a ‘story’ which is presented to FDA for review. The story must be consistent throughout and must ‘flow’ in a manner which clearly shows that your device is as safe and effective as the predicate device.
- Cost:
- At this time, the cost for a 510(k) is $19,870. If you apply for being a ‘small business’, the fee is reduced to $4,967. These fees typically go up every year.
- Timing:
- Acceptance review- Within 15 calendar days of receipt, FDA will conduct an Acceptance Review to determine whether your submission is complete and can be accepted for substantive review. If your submission has been found incomplete, within 15 calendar days FDA will notify you that the submission has not been accepted and identify those items that are the basis for the refuse to accept (RTA) decision and are therefore necessary for the submission to be considered accepted. The submission will be placed on hold and the 90 day review clock will not start until the missing elements are provided. When your submission is accepted for review, you will receive an ‘Acknowledgement Letter’ which has a ‘K’ number which will be used for your submission and clearance.
- Substantive Interaction- Once your submission has been accepted for review (i.e., after the RTA phase of review), FDA will conduct the substantive review and communicate with you within 60 calendar days of receipt of the accepted 510(k) submission. The communication can be an ‘Additional Information’ request or an email stating that FDA will continue to resolve any outstanding deficiencies via Interactive Review. If your submission is really good, the interaction may come in the form of a ‘Substantially Equivalent’ (SE) letter. This would mean that your submission was ‘cleared’ by FDA! receiving an SE letter as your initial communication is pretty rare but your chances of that occurring are increased by having presub meetings and by having someone on your team who is very familiar with creating ‘bullet-proof’ 510(k)s. We can help with that as we specialize in Software as A Medical Device (SaMD) submissions.
- Decision- FDA’s goal is to make a final determination for your submission by day 90 (remember, FDA’s clock stops if your submission gets put on hold). The determination will either be ‘Substantially Equivalent’ (SE) which means your device is cleared or ‘Not substantially Equivalent’ NSE which means your submission failed to prove that your device was as safe and effective as the predicate device. FDA typically lets you know that your submission is not going to get cleared (NSE determination). When this happens, you may request that the the submission be withdrawn. You do not get a refund of the review fee but you also do not have a ‘failed’ submission on record. If you have utilized the free presubmission meeting program, your chances of your device being cleared are higher as there should not be any surprises which could require significant—and expensive—remediation efforts.
Step 15: QMS Preparation and Marketing 🔗
- It is important to understand the limitations regarding medical devices which come to market through the 510(k) regulatory pathway. Here are a few items which will be helpful in staying on FDA’s good side:
- When you have submitted your 510(k) with FDA, you may display and advertise the device within the limits of the Intended Use and Indications which are stated in your submission.
- You may state that that there is a “510(k) pending” for the device. We advise that you also state; “not currently for sale in the United States.”
- While waiting for clearance, you may not take orders or be prepared to take orders which may result in a sale of the device.
- While we wait for FDA clearance, we need to start thinking about getting a Quality Management System (QMS) in place so we can legally sell our product after we receive FDA clearance.
- A QMS is needed to comply with FDA’s Quality System Regulation. Creating and implementing a QMS can be somewhat complicated and daunting if you don’t have experience. We have created a lean and efficient QMS which is specifically designed for micro and small businesses but which is scalable to any size business.
- Once you have a QMS in place, you will need to register your business (FDA calls this ‘Establishment Registration’) and list your device in the FDA’s database. Once you have your clearance, a QMS, Establishment Registration and Device listing, you can sell and distribute your device in the United States.
Step 16: FDA Clearance and Beyond 🔗
Congratulations you made it! Once your device is successfully on the market in the United States, you will need to follow the regulations related to such things as complaint handling, adverse event reporting and tracking where your devices are being used. You will also need to maintain your QMS as long as your device is on the market in the United States. FDA will inspect your business every few years to make sure that all regulations are being followed.
You must assess whether any modifications to the cleared device will require a new 510(k). Generally, you can retrain your AI on new data without needing to resubmit a 510(k). However, you must document the changes as if you were submitting one.