Introduction 🔗

De Novo requests provide a regulatory pathway for novel medical devices that are low to moderate risk.
This article covers best practices for preparing De Novo requests, frequently asked questions about the process, and examples of successful submissions. Emphasis is placed on diagnostic devices, my area of specialty. I hope the content is useful to startups or regulatory consultants who are seeking to bring products to market through the De Novo pathway.
About the Author 🔗

Hi, I’m David Giese, a Partner at Innolitics. I’ve helped bring 35+ diagnostic medical devices onto the US market. I take pride in the in-depth articles I write, and appreciate feedback and questions. I’d love to connect on LinkedIn, where I post pragmatic tips about medical-device regulations for over 5k followers.
Best Practices 🔗
- Engage the FDA early with a pre-sub specifically focused on the De Novo pathway. On some occasions, the FDA has pushed us to use the 510(k) pathway when we believed a De Novo was more appropriate.
- Also plan to do a pre-sub for your clinical study design.
- FDA typically requires more extensive testing and scientific evidence than for 510(k)s.
- When drafting special controls and benefit-risk determinations, review multiple other De Novos.
Basics 🔗
What is a De Novo Request? 🔗
A De Novo Request, also called a De Novo classification request, is a submission that is made to FDA for a novel medical device that meets these two criteria:
- It is believed to be class-I (lower risk) or class-II (moderate risk)
- No suitable predicate devices are on the market, so the 510(k) pathway isn’t available.
In Latin, “de novo” means “from the beginning”, which is appropriate since this pathway is the beginning of a new class of medical devices.
To do a De Novo request, the device should be sufficiently understood to explain all of its risks and benefits, such that all risks can be appropriately mitigated through general and/or special controls.
How are De Novo requests different from 510(k)s? 🔗
The purpose of a 510(k) is to show that a device is substantially equivalent to an existing legally-marketed device.
The purpose of a De Novo request is to justify why there are no predicate devices, but that the device should be class I or class II with specific special controls.
De Novo requests require clinical studies much more often than 510(k)s. However, not all De Novo requests require clinical data, and some 510(k)s do require clinical studies.
How much do De Novo requests cost? 🔗
De Novo requests are almost always more expensive than 510(k)s. My friend who works at FDA said they put about 2x the amount of review time and staff on De Novos, as compared to 510(k)s. This is consistent with our experiences on De Novo submissions, where FDA tends to provide very detailed, concrete feedback and thoroughly critiques your benefit-risk profile, marketing claims, and scientific evidence.
The key factors driving the cost include:
- Procuring scientifically valid evidence to justify your benefit-risk profile
- Team salaries during the longer timelines involved
- Cost for compiling and submitting the De Novo application
- FDA submission fee ($40,559 for small business and $162,235 otherwise in 2025)
- Regulatory support for one or more pre-subs
Also, here is a note from this 2022 survey:
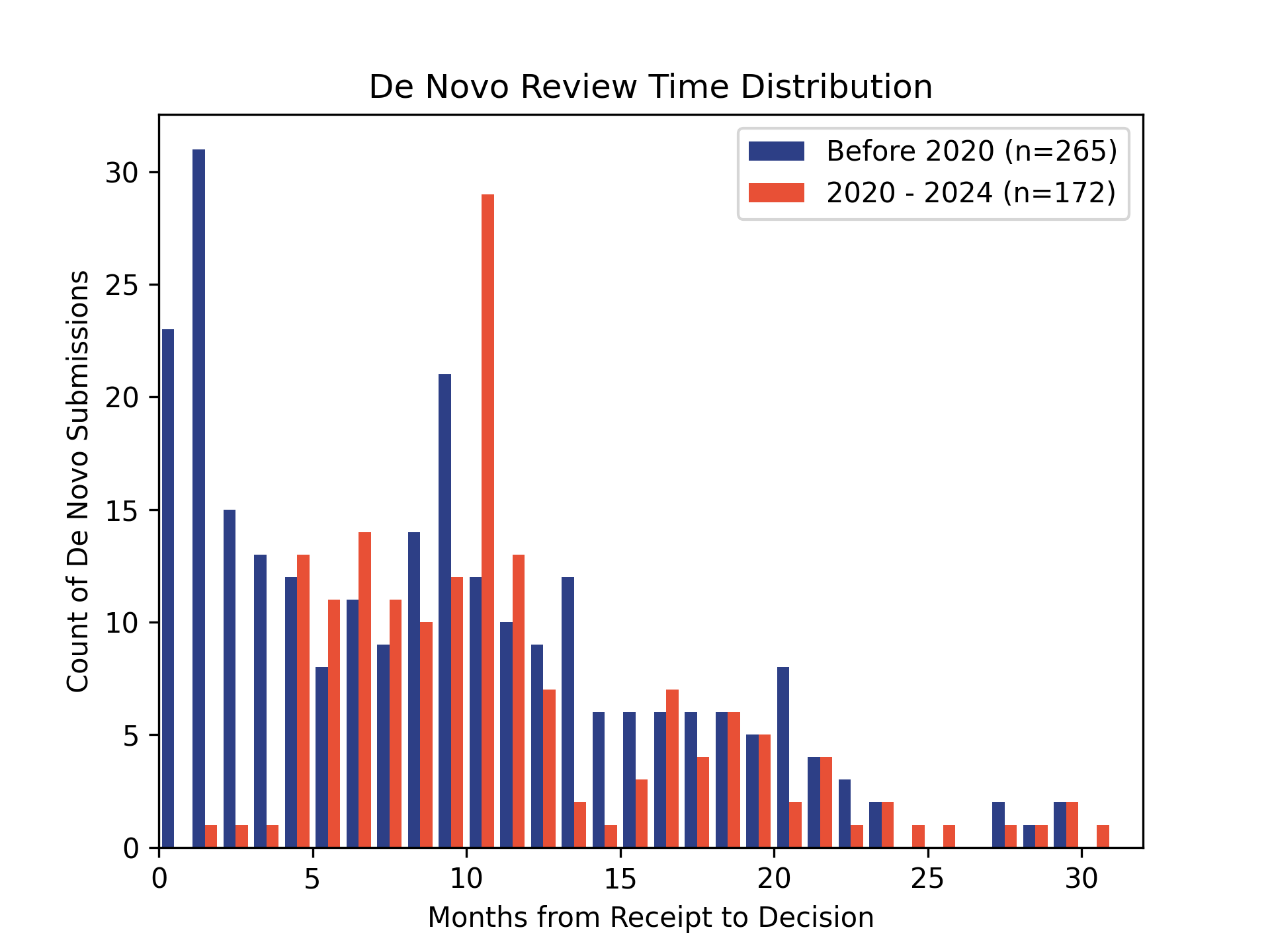
How long do De Novo requests take? 🔗
Although the FDA’s review clock is 150 days for De Novos, in practice the submission time is much longer to account for the time spent responding to FDA’s requests for additional information during the submission.
How often do De Novos get declined? 🔗
The FDA publicly tracks De Novo decisions in its database. Although many De Novos are eventually granted, rejections or withdrawals do occur (e.g., if the sponsor cannot demonstrate safety and effectiveness, or if the FDA determines the device should be Class III). Public data indicate that sponsors often need multiple rounds of additional information requests.
Each time a De Novo is submitted, it is assigned a new number. E.g., “DEN210052” is the 52nd De Novo in 2021. Since we can see all of the submissions that are granted, we can infer how many were NOT granted. While not systematic, this gives us a feeling. In 2021, approximately 51% of the De Novos were not granted.
Using an experienced team that understands your domain and has a strong regulatory, technical and clinical background will increase your chances of success.
How common are De Novo requests? 🔗
510(k) clearances are much more common than De Novos. The following tables and graphs were generated from the FDA’s publicly available databases. The number of De Novos is based on the year the De Novo decision was granted.
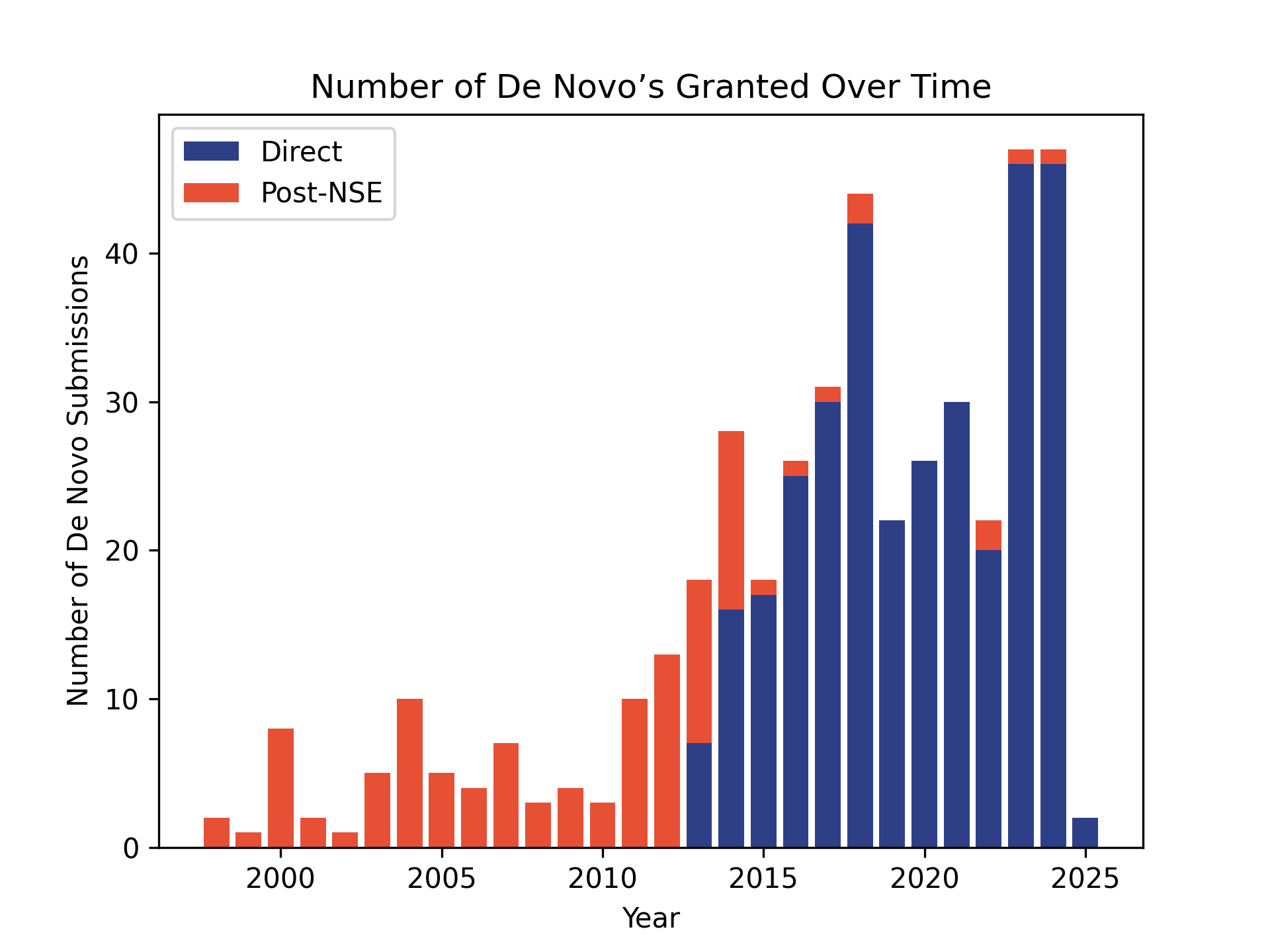
| Year | De Novo Requests | 510(k) Submissions |
|---|---|---|
| 2024 | 47 | 3,083 |
| 2023 | 47 | 3,301 |
| 2022 | 22 | 3,195 |
| 2021 | 30 | 2,995 |
| 2020 | 26 | 2,901 |
| 2019 | 22 | 2,896 |
| 2018 | 44 | 3,020 |
| 2017 | 31 | 3,174 |
| 2016 | 26 | 2,930 |
| 2015 | 18 | 3,006 |
| 2014 | 28 | 3,175 |
| 2013 | 18 | 3,034 |
| 2012 | 13 | 3,123 |
| 2011 | 10 | 3,115 |
| 2010 | 3 | 2,804 |
| 2009 | 4 | 3,004 |
| 2008 | 3 | 3,050 |
If a De Novo is granted, can others use it as a predicate later? 🔗
Yes. Once the De Novo is granted, a new classification regulation is established, and the device can become a predicate for future 510(k)s provided the new device falls under that same classification regulation and meets the same special controls. This is part of the reason De Novos effectively “open the path” for follow-on devices.
One way companies protect themselves from follow-on submissions is by proposing special controls that require thorough clinical testing, effectively raising the bar for future entrants.
What types of devices have De Novos? 🔗
The following table shows the distribution of De Novos across medical review panels. Relatively few have been granted in radiology.
| Panel Name | 2020-2024 | Before 2020 |
|---|---|---|
| Gastroenterology & Urology | 19 | 22 |
| Cardiovascular | 18 | 16 |
| Microbiology | 16 | 42 |
| General Hospital | 14 | 10 |
| Neurology | 14 | 28 |
| Orthopedic | 14 | 2 |
| General & Plastic Surgery | 13 | 30 |
| Radiology | 9 | 7 |
| Ear, Nose, & Throat | 8 | 8 |
| Ophthalmic | 8 | 10 |
| Anesthesiology | 7 | 7 |
| Clinical Chemistry | 7 | 19 |
| Immunology | 5 | 21 |
| Hematology | 5 | 4 |
| Dental | 4 | 4 |
| Pathology | 4 | 5 |
| Obstetrics/Gynecology | 2 | 13 |
| Clinical Toxicology | 2 | 7 |
| Physical Medicine | 1 | 5 |
| Molecular and Clinical Genetics | 1 | 5 |
What are the benefits of pursuing a De Novo? 🔗

For many devices and intended uses, there is no choice. Either you’re a 510(k) or a De Novo. However, there are gray areas where you can lean one way or another and, of course, given two alternative devices and intended uses where one is a 510(k) and another is a De Novo, a founder may need to weight the benefits of the De Novo pathway. There are a couple benefits to a De Novo:
- It can help with fund-raising. Founders will often pitch investors on their device's groundbreaking innovation while simultaneously planning to pursue a 510(k) pathway. Sophisticated investors know that a 510(k) clearance is based on “substantial equivalence”, and thus if a 510(k) will work, there can be limited novelty. The De Novo pathway demonstrates to investors that your device is genuinely novel.
- A second benefit of the De Novo pathway is that you can influence the special controls for your device regulation. All future devices will need to follow these special controls as well, therefore, you can try to use them to create a competitive moat. E.g., picking special controls that dove tail with your intellectual property or are relatively easier for your company to complete.
What are the downsides of a De Novo? 🔗

The timelines are longer and in many cases you’ll require more clinical evidence.
I’ve heard from an ex-FDA reviewer that the FDA devotes about twice as much time to the review of a De Novo as compared to a 510(k). It is therefore reasonable to expect the review to be more thorough and in-depth.
On top of all of this, subsequent devices will be able to “follow in your footsteps” using the less burdensome 510(k) pathway.
Process 🔗
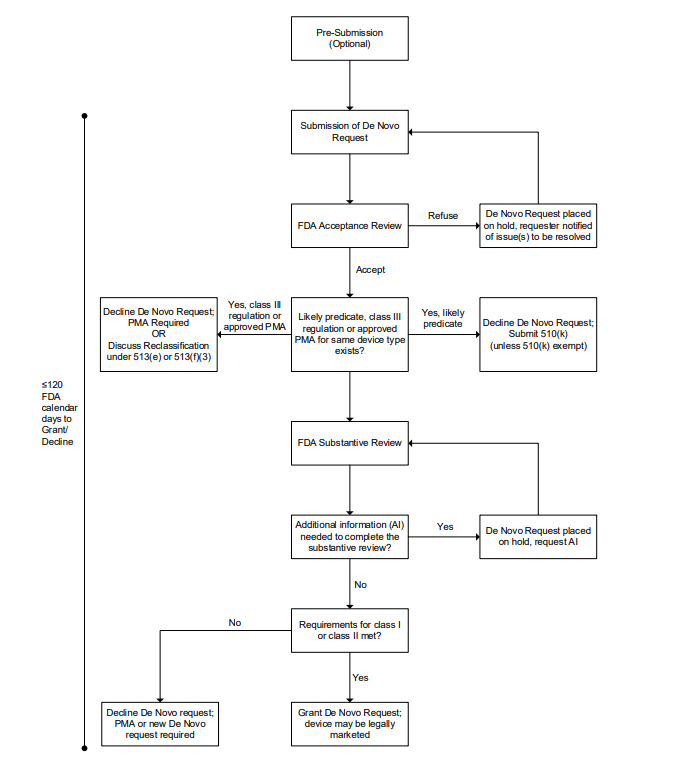
What is the process for completing a De Novo request? 🔗
- Complete a Pre-Sub (optional, but highly recommended)
- Compile the De Novo submission into the eSTAR
- Submit to FDA
- Acceptance Review (up to 15 days)
- Substantive Review
- FDA completes initial review (up to 150 days)
- FDA inspects client’s QMS (we personally have not encountered this in practice)
- Manufacturer revise or provide additional information (optional)
- FDA typically requests additional information
- Company responds to request for additional information (up to 180 days)
- FDA grants or declines the De Novo request
Just as is the case with a 510(k), you still need to register your facility and list the device with the FDA as a separate step.

What specific content is required for a De Novo vs a 510(k)? 🔗
A De Novo request will NOT contain these 510(k) sections:
- References to predicates and reference devices
- A substantial equivalence comparison
- Special controls adherence
A De Novo request contains other sections that aren’t in a 510(k):
- Classification
- Proposed classification (class I or class II)
- Justification for the classification
- Proposed classification name (e.g., “Radiological Computer-Assisted Triage And Notification Software”)
- Benefits, Risks, and Mitigation Measures
- Risk/Mitigation Table (note this is higher level and more abstract ISO 14971 risk table)
- Proposed special controls (see the bottom for examples)
- Alternative practices and procedures
A lot of the content between the 510(k) and De Novo is the same:
- Electromagnetic Compatibility (EMC)
- Wireless Technology
- Electrical, Mechanical, and Thermal (EMT) Safety Summary
- Reprocessing
- Sterility
- Labeling
- Shelf-life
- Biocompatibility
- Software documentation
- Documentation Level Evaluation
- Software Description
- Risk Management Plan
- Risk Assessment
- Risk Management Report
- Software Requirements Specification
- System and Software Architecture Design
- Software Design Specification
- Software Development, Configuration Management, and Maintenance Practices
- Software Testing as part of Verification and Validation
- Software Version History
- Unresolved Software Anomalies
- SOUP (OTS) Report
- Cybersecurity documentation
- Security Risk Management Report
- Threat Model
- Security Risk Assessment
- Software Bill of Materials (SBOM)
- Assessment of Unresolved Security Anomalies
- Cybersecurity Metrics
- Security Architecture Views
- Cybersecurity Controls
- Cybersecurity Testing
- Cybersecurity Labeling
- Cybersecurity Management Plan
What is the purpose of the Acceptance Review? 🔗
During the acceptance review, FDA checks if the submission contains all the required elements and information needed for a substantive review. This includes verifying that all sections are complete, appropriate test data is included, and the format follows FDA guidelines. If the submission is incomplete or inadequate, FDA will notify the requester and identify the missing elements that need to be addressed.
Acceptance review is meant to encourage quality applications from De Novo requesters and to allow the FDA to focus on complete applications.
How in-depth is the Substantiative Review? 🔗
In my experience, De Novo reviews are significantly more in-depth than 510(k) reviews. In the case of 510(k)s, the reviewers tend to focus on substantial equivalence. In the case of a De Novo, the reviewers take a more wholistic approach to the device.
What are proposed special controls? 🔗
Special controls are specific requirements that FDA establishes for class II devices to provide reasonable assurance of safety and effectiveness. When submitting a De Novo request, manufacturers must propose appropriate special controls that will help mitigate the identified risks of their device.
What are some examples of proposed special controls? 🔗
See the examples at the bottom of the page for special controls for particular devices. Here are some general categories of controls:
- performance standards
- performance testing
- postmarket surveillance
- patient registries
- development and dissemination of guidelines
- required clinical data to be included in future submissions
- labeling requirements
- design specifications.
What would be the purpose of the Pre-Sub? 🔗
FDA Strongly recommends doing a Pre-Sub prior to a De Novo submission, although it is optional (If you’re not familiar with Pre-Subs, checkout my article FDA Pre-Subs: Best Practices, FAQs, and Examples.)
The purpose of the pre-sub would be to
- Confirm FDA agrees the device requires a De Novo
- Present the identified “risks to health” for the device type
- Get feedback on your proposed special controls
- Get feedback on your proposed non-clinical and clinical study protocols
FDA has requested that Pre-Subs for De Novo submissions include:
- Device Description
- Intended use / indications for use
- Regulatory history
- Proposed class (I or II)
- Justification for why general and special controls are sufficient
- The 510(k) database searches you performed to rule out a 510(k)
- A list of relevant classification regulations
- As appropriate for the Pre-Sub:
- A breakdown of each identified risk to health and details of any studies needed to support the risk profile.
- Types of scientific evidence you plan to provide with the De Novo request.
- Protocols for non-clinical and clinical studies, showing how they address identified risks and demonstrate performance levels
- Proposed risk mitigation measures, clearly distinguishing between general and special controls
What happens if a similar De Novo request is granted while ours is in review? 🔗
If this were to happen, your request would be declined and you would then need to submit a new 510(k) submission using the other device as your predicate. What is worse, you would need comply with the new special controls defined by the other device. I would like to think FDA would be flexible regarding any clinical evidence you had collected, but the worst case scenario would be:
- You complete an expensive clinical study A
- Another De Novo is granted
- Study A doesn’t comply with new special controls
- You need to complete another expensive study B
Where can I learn more about De Novo requests? 🔗
- FDA Guidance:
- 2021 FDA Guidance - De Novo Classification Process (Evaluation of Automatic Class III Designation)
- 2021 FDA Guidance - Acceptance Review for De Novo Classification Requests
- 2019 FDA Guidance - Factors to Consider When Making Benefit-Risk Determinations in Medical Device Premarket Approval and De Novo Classifications
- Regulations
Examples 🔗
QuantX: The first CADx cancer detection 🔗
Introduction 🔗
I helped one of my talented clients clear another device in the POK product code in 2024. You can view it in our FDA browser here. This was possible because, back in 2017, FDA granted a De Novo request to Quantitative Insights, Inc., for their QuantX device.
By granting this De Novo request, FDA was agreeing to the following propositions:
- There were not devices on the market that were substantially equivalent.
- There was sufficient clinical evidence to believe it is a Class II device with the proposed special controls.
General Description 🔗
This established the 21 CFR 892.2060 regulation, which has the following general description:
Indications for Use 🔗
The QuantX device has a much more specific indications for use:
Identified Risks to Health and Mitigation Measures 🔗
As part of their submission, they identified the following “Risks to Health”:
| Identified Risks to Health | Mitigation Measures |
|---|---|
| Incorrect lesion(s) characterization leading to false positive results may result in incorrect patient management with possible adverse effects such as unnecessary treatment, unnecessary additional medical imaging and/or unnecessary additional diagnostic workup such as biopsy. | • Certain design verification and validation activities identified in special control (1) • Certain labeling information identified in special control (2) |
| Incorrect lesion(s) characterization leading to false negative results may lead to complications, including incorrect diagnosis and delay in disease management. | • Certain design verification and validation activities identified in special control (1) • Certain labeling information identified in special control (2) |
| The device could be misused to analyze images from an unintended patient population or on images acquired with incompatible imaging hardware or incompatible image acquisition parameters, leading to inappropriate diagnostic information being displayed to the user. | • Certain design verification and validation activities identified in special control (1) • Certain labeling information identified in special control (2) |
| Device failure could lead to the absence of results, delay of results or incorrect results, which could likewise lead to inaccurate patient assessment. | • Certain design verification and validation activities identified in special control (1) • Certain labeling information identified in special control (2) |
Special Controls 🔗
The FDA agrees that the following special controls (along with the general controls like the quality system regulation, device listing, etc.) will assure the safety and effectiveness of the device. Note that these special controls require all subsequent predicate devices to complete a clinical study to evaluate the reader performance. Although a reader study may very well be appropriate in this case, it is a common technique in De Novos for the submitting company to attempt to “defend” easy “me too” devices by suggesting special controls that require significant investment:
- Design verification and validation must include:
- A detailed description of the image analysis algorithms including, but not limited to, a detailed description of the algorithm inputs and outputs, each major component or block, and algorithm limitations.
- A detailed description of pre-specified performance testing protocols and dataset(s) used to assess whether the device will improve reader performance as intended.
- Results from performance testing protocols that demonstrate that the device improves reader performance in the intended use population when used in accordance with the instructions for use. The performance assessment must be based on appropriate diagnostic accuracy measures (e.g., receiver operator characteristic plot, sensitivity, specificity, predictive value, and diagnostic likelihood ratio). The test dataset must contain sufficient numbers of cases from important cohorts (e.g., subsets defined by clinically relevant confounders, effect modifiers, concomitant diseases, and subsets defined by image acquisition characteristics) such that the performance estimates and confidence intervals of the device for these individual subsets can be characterized for the intended use population and imaging equipment.
- Standalone performance testing protocols and results of the device.
- Appropriate software documentation (e.g., device hazard analysis; software requirements specification document; software design specification document; traceability analysis; description of verification and validation activities including system level test protocol, pass/fail criteria, results, and cybersecurity).
- Labeling must include:
- A detailed description of the patient population for which the device is indicated for use.
- A detailed description of the intended reading protocol.
- A detailed description of the intended user and recommended user training.
- A detailed description of the device inputs and outputs.
- A detailed description of compatible imaging hardware and imaging protocols.
- Warnings, precautions, and limitations, including situations in which the device may fail or may not operate at its expected performance level (e.g., poor image quality or for certain subpopulations), as applicable.
Here are links to the original classification letter and the decision summary.
Viz.ai: Cardiovascular machine learning-based notification software 🔗
Introduction 🔗
Many of my projects are in imaging, but I’ve also worked on a few diagnostic devices that operate on temporal data, such as that from ECGs.
The Viz HCM De Novo was granted on August 3rd, 2023. It only took 8 months. It established the 21 CFR 870.2380 regulation and the QXO product code. You can view it in our FDA browser. Unlike the POK product code, this one doesn’t yet have any devices that have used it as a predicate. Here is the general description:
Identified Risks to Health and Mitigation Measures 🔗
| Identified Risks to Health | Mitigation Measures |
|---|---|
| False positive or false negative leading to incorrect treatment or diagnosis | • Clinical performance testing • Non-clinical performance testing • Labeling |
| Incorrect treatment or diagnosis due to model bias or failure to adequately generalize to the intended use population | • Clinical performance testing • Labeling |
| Device used in unsupported patient population or with unsupported input/hardware | • Labeling • Human factors assessment • Software verification, validation, and hazard analysis |
| Overreliance on device output for follow-up | • Human factors assessment • Labeling |
Special Controls 🔗
- Clinical performance testing must demonstrate that the device performs as intended under anticipated conditions of use. The following must be met:
- Clinical validation must use a test dataset of real-world data acquired from a representative patient population. Data must be representative of the range of data sources and data quality likely to be encountered in the intended use population and relevant use conditions in the intended use environment. The test dataset must be independent from data used in training/development and contain sufficient numbers of cases from important cohorts (e.g., demographic populations, subsets defined by clinically relevant confounders, comorbidities, and subsets defined by hardware and acquisition characteristics) such that the performance estimates and confidence intervals of the device for these individual subsets can be characterized for the intended use population and acquisition systems (e.g., acquisition hardware or preprocessing software). Study protocols must include a description of the adjudication process(es) for determining ground truth of training and test datasets;
- Data must be provided within the clinical validation study or using equivalent datasets to demonstrate the consistency of the output over the full range of inputs;
- Performance goals used to determine success of clinical validation must be justified in the context of risks associated with follow-up testing;
- Objective performance measures (e.g., sensitivity, specificity, positive predictive value or negative predictive value) must be reported with relevant descriptive or developmental performance measures. Summary level demographic information and sub-group analyses must be provided for each study site, relevant demographic sub-groups, and acquisition systems; and
- The test dataset must include a minimum of 3 geographically diverse sites, separate from sites used in training of the model.
- Software verification, validation, and hazard analysis must be performed. Software documentation must include:
- A description of the model/algorithm, algorithm inputs/outputs, and supported patient population;
- Integration testing in the intended software system or software environment; and
- A description of the expected impact of all applicable sensor acquisition hardware characteristics on performance and any associated hardware specifications, including:
- A description of input signal / data quality control measures; and
- A description of all mitigations for user error or failure of any subsystem components (including signal detection, signal analysis, data display, and storage) on output accuracy.
- Human factors assessment of the intended users in the intended use environment must evaluate the risk of misinterpretation of device output.
- Labeling must include:
- A summary of the performance testing methods, tested hardware, tested/supported patient population, results of the performance testing for tested performance measures/metrics, summary-level descriptions of patient demographics and associated subgroup analyses for training and test datasets, and the expected minimum performance of the device;
- Device limitations or subpopulations for which the device may not perform as expected;
- Warning that the user should not rely on the lack of a suspected finding to rule-out follow-up;
- A statement that the device output should not replace a full clinical evaluation of the patient and that the output may not be sufficient as the sole basis for further testing;
- Warnings identifying sensor acquisition factors that may impact measurement results;
- Guidance for interpretation of the measurements and typical follow-up testing; and
- The type(s) of hardware sensor data used, including specification of compatible sensors for data acquisition.
Revision History 🔗
| Date | Changes |
|---|---|
| 2025-01-28 | Initial Version |
| 2025-02-12 | Added questions about pre-subs for De Novos and a discussion of what happens if multiple De Novos are submitted. |
| 2025-05-24 | Added questions about the benefits and problems with pursuing a De Novo. |