


Introduction 🔗
If you’re bringing new medical-device software to the United States market, then this guide is for you. We begin with best practices that are useful early in your journey. Then we provide succinct answers to questions we’re commonly asked by our clients. Finally, we list some detailed examples.
The focus is big-picture—costs, timelines, overall process, and project risks. Our answers target Software as a Medical Device (SaMD), although much will apply to devices generally.
If you have other questions, please ask using the chat button in the bottom right corner.
Thank you Jim Luker and Mary Vater for reviewing this article and providing feedback and additional insights.
About the Author 🔗

Hi, I’m David Giese, a Partner at Innolitics. I’ve wrote this article based on my real-world experiences bringing 35+ diagnostic medical devices onto the US market. I take pride in the in-depth articles I write, and appreciate feedback and questions. I’d love to connect on LinkedIn, where I post pragmatic tips about medical-device strategy, pre-subs, and regulations for my >8k followers.
Best Practices 🔗
Here are best-practices that we’ve observed by working with our clients over the years:
- Establish a regulatory strategy early. Once you can describe your device’s functionality and intended use, you should establish and document a regulatory strategy. This should include the necessary clinical testing, non-clinical testing, software documentation, and cybersecurity testing. Not all devices need all of these, but if yours does, each by itself can add $100,000s of cost and months or years of delays to your projects. It’s best to identify them as early as possible so you don’t set unrealistic investor expectations.
- If your product is SaMD, pick a regulatory team that knows software and cybersecurity. This is especially true if you plan to make significant changes to the software after the first release or have flexibility with regards to what features are included in the initial release. SaMD is significantly different than hardware devices and there are more strategic options available.
- Take regulatory risks that align with business needs. Many regulatory professionals not only attempt to avoid all FDA deficiencies but also over-emphasize how stringent the FDA will be. For most startups, the goal isn’t an FDA submission that flies past the FDA without any pushback; if it does, it may mean you went overboard with documentation or clinical-evidence gathering and lost valuable time getting your device to market. Instead, the goal is to build a safe and effective device, and then to get it through FDA as quickly and cheaply as possible.
- Hire software engineers who have worked on medical devices before. If they haven’t, it will be expensive and time-consuming for them to write the documentation needed for FDA regulatory submissions. We routinely see it take software teams 9 - 12 months to create the documentation if they haven’t done it before.
- Plan your cybersecurity controls early in the development process. FDA puts a strong emphasis on cybersecurity. You will need to implement a security risk management process and a number of cybersecurity controls, and it’s easiest to do so if you plan for them instead of trying to insert them into the software after the fact.
- Don’t document too much before the design is settled. Most medical device startups set up their Design Controls (part of a Quality Management System) process too late, causing documentation busy work and engineering rework. On the other hand, it is also possible to begin documenting too much too early. Documenting your plan and high-level needs and risks early is valuable, but much of the lower-level documentation is best done later in the process when the design has settled and the chances of re-work are lower.
Device Classification 🔗
Who regulates medical devices in the US? 🔗
The Food and Drug Administration (FDA) is the primary regulatory body that oversees medical devices in the United States.
Can software be considered a medical “device”? 🔗
Yes! The word “device” confuses people because in everyday language it implies a physical thing. In regulatory language, the term “device” includes software. Software-only devices are referred to as “Software as a Medical Device”, or SaMD. Software that is part of a medical device is called “Software in a Medical Device”, or SiMD.
The company that creates the software is the “manufacturer”.
When is software considered a medical device? 🔗
Software that is “intended for use in the diagnosis of disease or other conditions, or in the cure, mitigation, treatment, or prevention of disease” is considered a medical device. Some devices are considered “general wellness” devices and aren’t actively regulated.
Note that what makes a product a medical device is not just its technical capabilities, but also its intended use. The intended use is determined from its labeling, including the instructions for use and the marketing claims you make about the device (e.g., on your website).
Usually, it is clear if software is a device. If it’s not clear, FDA’s How to Determine if Your Product is a Medical Device page is a good starting place. If you’re still not sure, contact us or another regulatory consulting agency for help.
What regulatory controls do medical-device manufacturers need to follow? 🔗
In the United States, medical devices fall into three classes according to their risk level:
| Class | Risk Level | Regulatory Controls |
|---|---|---|
| 1 | Lowest | General Controls |
| 2 | Moderate | General Controls and Special Controls |
| 3 | Highest | General Controls and Premarket Approval (PMA) |
All devices must follow general controls unless exempted by regulations. If a device is exempted from one of the general controls, such exemption is stated in the classification regulation for that device. Class II devices often will have special controls that apply to just that category of product. Class III devices must undergo Premarket Approval (PMA), the most rigorous market pathway.
How do you determine a device’s safety class? 🔗
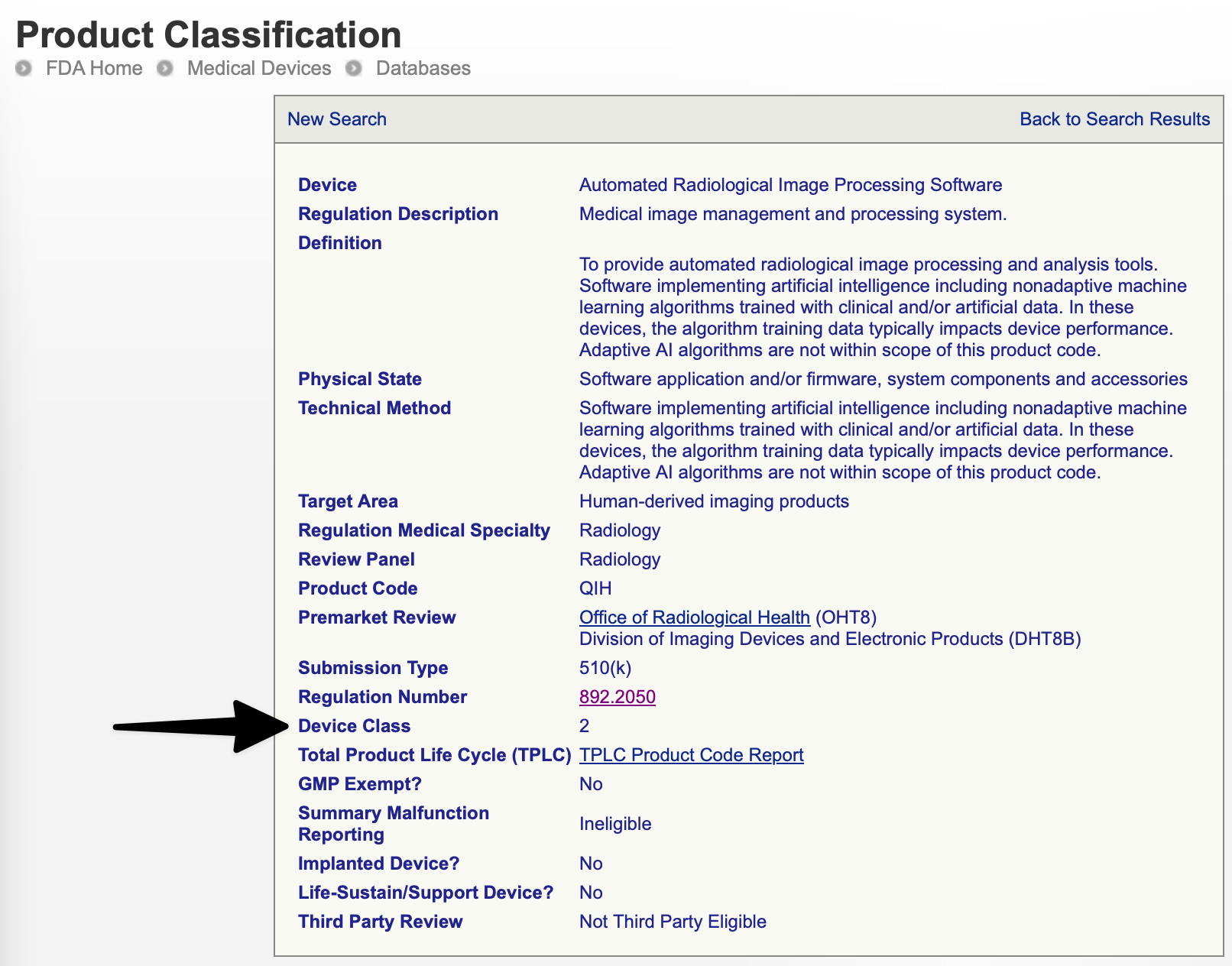
The FDA classifies and tracks types of devices using three-letter product codes. E.g., "LLZ" is the product code for "Medical image management and processing systems.” (See the examples section below for many more.)
Each product code traces back to a Regulation Number. (Some regulation numbers may have many product codes, but some have a single product code.)
Once you’ve looked up a device’s product code (see next question), you can look up its regulation, and in turn its safety class and applicable special controls.
Note that some devices may have multiple product codes if there were multiple predicates.
How do you look up a device’s product code? 🔗
If you know of similar devices that are on the market, you may look up their product code in the FDA’s 510(k) database.
If you’re not aware of similar devices, you may also browse all of the product codes and their descriptions in our FDA database browser, which is a tool built off of the FDA’s product code database. Product codes are organized by review panel (e.g., Radiology, Cardiology, Pathology, etc.).
What if your device doesn’t have a product code? 🔗
If your low or medium risk device doesn’t fit well into any existing product codes, you may need to submit a De Novo Classification request. This is a somewhat more involved process than if you do have a product code. You can read more about it here. We can help with De Novo Classification requests.
FDA also has another method for determining the product code and regulatory requirements for a device that is novel or which does not fit well into existing product codes. This method is called a 513(g) Request for Information. With this type of request, you provide FDA with detailed information for your software and in return they provide a detailed document which includes the safety class as well as the regulatory requirements for the device. It is also possible that they will tell you that your software does not fit the definition of a medical device and therefore is not regulated in the United States at this time. 513(g) requests have an associated fee ($3264 for a small business—typically a business with less than $100 million in gross receipts). Unless you are really stuck, we suggest you use the other available free resources (i.e., FDA’s Web Site our FDA Browser) to determine the regulatory requirements for your software.
What are general controls? 🔗
Software devices in all three classes require you to follow these general controls before placing your device on the market in the United States. General controls include
- Registering your organization with the FDA and list your devices (easy)
- Avoiding misleading statements about your device’s efficacy, safety, or intended use, also known as “misbranding” (easy, but can make marketing difficult)
- Setting up and maintaining a Quality Management System (time consuming)
- Reporting deaths, serious injuries and malfunctions, etc. (hopefully you never have to do this, but procedurally, it’s easy to do)
- Unique Device Identification (UDI) labelling requirements
There are a few additional controls that are more relevant for hardware devices.
Note also that 510(k) premarket notifications are technically a general control, however most class I devices are “exempt” from this general control. Said another way, most Class I devices are exempt from doing 510(k)s. We’ll discuss 510(k) submissions later.
What are special controls? 🔗
Special controls are additional regulatory requirements for specific groups of class II devices. You can see some examples of special controls at the bottom of this article. Typical special controls include:
- Performance standards
- Postmarket surveillance
- Patient registries
- Special labeling requirements
- Premarket data requirements
- Guidelines
How do you look up the special controls that apply to a particular device? 🔗
Once you’ve identified the product code for your device, you can find what regulation number applies to it. When you open the regulation within the Code of Federal Regulations (CFR), you will see if there are any special controls for that device. Please note that there may be multiple product codes for a particular regulation number.
For example, the 892.2050 regulation has 11 product codes. When you open up the 892.2050 regulation you will see that the special controls are: voluntary standards - Digital Imaging and Communications in Medicine (DICOM) Std., Joint Photographic Experts Group (JPEG) Std., Society of Motion Picture and Television Engineers (SMPTE) Test Pattern).
You can also see the regulation text in our FDA database browser.
Sometimes, the special controls are not listed under the regulation. In the case where a De Novo was recently granted, and a new product code created, the source of the Special Controls would actually be in the De Novo reclassification order, available through the De Novo Database. You can search by your product code to see if there was a De Novo granted, and to see if it lists special controls.
One example of this situation is product code QVD defined as: Radiological Machine Learning Based Quantitative Imaging Software With Change Control Plan. The De Novo was granted in Feb. 2023, but there is not yet a regulation record on the FDA website, and Special Controls are only identified in the De Novo Reclassification Order.
As mentioned above, sometimes there are multiple product codes listed under a single regulation, and there may be special controls for some, but not all of the product codes. In that case, the special controls again may not be listed in the regulation.
You should also search the FDA Special Controls Database. Again, unfortunately, not all special controls are listed there as released as formal guidance documents, but it is another potential source of this information.
It is recommended that you check in the multiple places where special controls may be identified before determining that there are none.
What is a premarket submission? 🔗
A premarket submission is a set of documents that you send to the FDA, which they use to evaluate whether your device is safe and effective prior to allowing you to market the device in the United States.
| Class | Risk Level | Premarket Submission |
|---|---|---|
| 1 | Lowest risk | Typically not needed |
| 2 | Moderate risk | Typically a 510(k) or De Novo |
| 3 | Highest risk | PMA |
Most SaMD is Class 2. You can read about the difference between these premarket submissions in the later sections of this document.
Note that most Class 1 and some Class 2 devices are exempt from requiring a 510(k) submission.
What is a the difference between a 510(k) and a De Novo? 🔗
A 510(k) submission aims to show your device is substantially equivalent to an existing device, called the predicate device. Substantially equivalent means that your device and the predicate device(s) have the same intended use, and technological characteristics. Your device may have some differences relating to technological characteristics but they must not raise different questions regarding safety and/or effectiveness. At times, you may compare your device to more than one device (multiple predicates). A De Novo request is a request for FDA to classify your device, because you believe that there are no suitable predicates and that it is not Class 3 (highest risk).
Do I need to submit a 510(k)? 🔗
You typically need to make a 510(k) submission if all of the following are true:
- You want to sell your product in the United States.
- Your product is considered a medical device by the FDA.
- Your product is classified by the FDA as a non-exempt class 2 device. Rarely, a class 1 device may require a 510(k).
- Your product is “substantially equivalent” to an existing medical device (or devices).
This is somewhat oversimplified, and there are many exceptions to this process. To truly determine if you need a 510(k), we suggest having a regulatory consultant create a regulatory strategy for your product idea.
What is a regulatory strategy? 🔗
The specific content of a regulatory strategy can vary, but the fundamental questions we try to answer for our clients in a regulatory strategy are as follows:
- Business goals (short term and long term)
- Is it a medical device based on it’s device description and intended use?
- How is it classified in particular jurisdictions (i.e., United States, EU)?
- What are the available regulatory pathways (e.g., 510(k) or De Novo)?
- What are the expected regulatory expenses and timelines for the selected regulatory pathway?
- What clinical validation or performance testing will be necessary?
- Should we engage with FDA in a pre-sub (a formal, one-hour meeting with FDA)?
- What are the largest regulatory risks?
- If there’s flexibility, which software features should we include in our device first?
- How will certain features affect the regulatory strategy?
- What are the overall costs and timelines we can expect?
You can read more about our regulatory strategy service here.
When do I need to follow all of these controls? 🔗
Startups who are starting from nothing will interact with FDA following a timeline that looks like this:
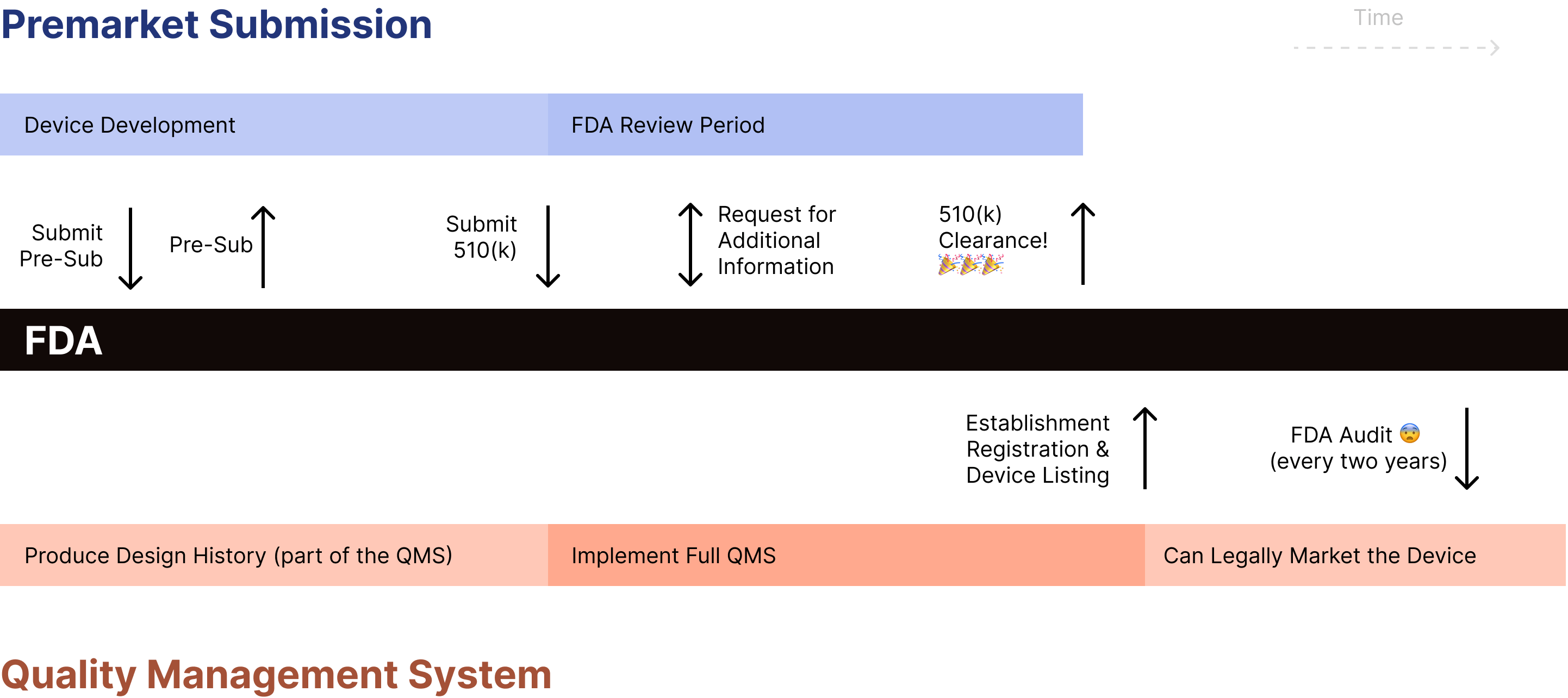
Note that there are two “swim lanes”. The top swim lane is the premarket submission (usually a 510(k)). The bottom swim lane is the quality management system. The two swim lanes interact; many of the documents in the design history file will be part of the 510(k) but are also controlled documents in your QMS. Many startups don’t begin fully executing their QMS until they’ve received 510(k) clearance, however, it’s a good idea to begin implementing it sooner.
510(k) Premarket Notification 🔗
What is a 510(k)? 🔗
A 510(k) is the most widely used pathway for putting medium-risk medical-device product onto the US market. As previously mentioned, it involves showing that your device is substantially equivalent to an existing medical device that has been ‘cleared’ through the 510(k) or De Novo pathway. This existing device is called the predicate device.
In a 510(k), you’re showing your device equivalent to the previously cleared device(s) and is thus as safe, secure and effective. In a De Novo request or a PMA, you’re showing that the device is safe, secure, and effective on its own merits.
What does substantial equivalence mean? 🔗
Substantial equivalence means that the new device is as safe, secure, and effective as the predicate device.
A device is substantially equivalent if, in comparison to a predicate, it
- has the same intended use as the predicate, and it
- has the same technological characteristics as the predicate.
Alternatively, if there are differing technological characteristics, substantial equivalence can be shown if it
- has the same intended use as the predicate, and it
- has different technological characteristics that do not raise different questions of safety and effectiveness, and
- the information submitted to FDA demonstrates that the device is as safe and effective as the legally marketed device.
A claim of substantial equivalence does not mean the new and predicate devices need to be identical.
This can be visualized in the flow-chart from the FDA guidance (which absolutely should be consulted in detail when making these decisions—the flow chart is not intended to be used “stand-alone”.
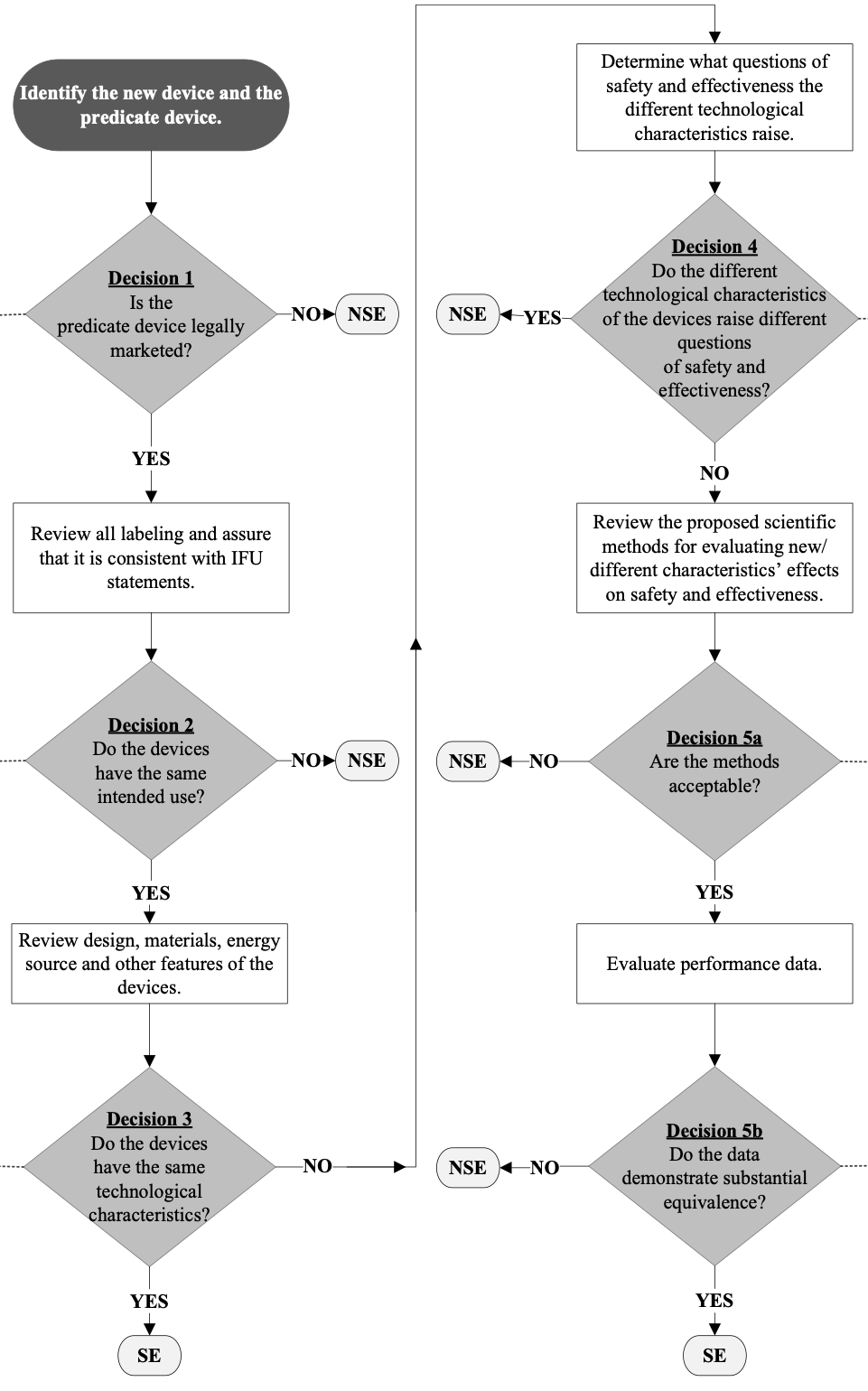
As you can imagine, substantial equivalence is a bit of a gray area. Often, medical device startups have something new they want to bring to market, yet they also want to use the less burdensome 510(k) regulatory pathway. As a result, it is not uncommon to stretch the concept of substantial equivalence. If you’re doing this, you’ll want to think through the comparison carefully, since if FDA disagrees, they will reject your 510(k) and issue a ‘Not Substantially Equivalent (NSE) decision. If you receive an NSE determination, you may not market your device in the United States and will need to create and submit another 510(k).
What is the process for submitting a 510(k)? 🔗
At a very high level you need to:
- Create a lot of documentation following a specific format
- Pay the submission fee
- Submit the documentation to FDA
- Wait (usually 60 days)
- Receive an additional information request from FDA (180 days response timeframe)
- Answer FDA’s request for additional information
- FDA decides if your device is “Substantially Equivalent” or “Not Substantially Equivalent”
- If the device is Substantially Equivalent, you can begin marketing the device.
You still need to fulfill the other general regulatory requirements, as spelled out in the “Post Market” section of this article. In general, it is a good idea to understand and begin preparing for the general controls while your 510(k) submission is under review.
What documentation goes into a 510(k)? 🔗
At a high level, the documentation in a 510(k) includes
- The indications for use and intended use of your software
- A detailed description of how your software is “substantially equivalent” to a previously cleared medical device software (also known as your “predicate device”)
- A detailed description of how your software works (including architectural diagram(s))
- A detailed description of your cybersecurity risks and controls
- A detailed description of how your software can directly or indirectly harm patients
- A detailed description of how you’ve mitigated these risks
- A detailed description of how you’ve ensured your software works properly (verification and validation).
You then format the documents in a specific way that makes it easy for the FDA to review, send it to them, respond to their questions, and then hopefully get a letter stating that you can now sell your software in the US.
You may want to check out our Ultimate 510(k) Checklist to see a granular list of what goes into a 510(k) for SaMD devices.
It’s not uncommon for the submissions to run over 1,000 pages in length.
What do we do if there aren’t any existing medical devices like ours? 🔗
First of all, the concept of “substantial equivalence” can be tricky. Unless your device is truly unique it’s likely there are existing devices on the market that are similar. In some cases you can even have multiple predicate devices. That said, you don’t want to guess here. Many 510(k)s are rejected, and years of people’s lives wasted, along with millions of dollars, because the FDA disagrees with your assessment of what is equivalent.
If it is determined that there are no equivalent devices on the market in the United States, you will not be able to use the 510(k) pathway and will need to consider the other available options. As most software medical devices are class 2 (medium-risk), the De Novo request process may be applicable. A De Novo is similar to a 510(k) but the device must stand on its own in terms of safety, security and effectiveness. De Novo requests often require some level of clinical validation testing. If your software happens to be class 3 (high-risk), you will need to create and submit a PMA. This is the most stringent type of submission. We can help with all types of regulatory submissions.
How long does it take to put together a 510(k) Submission? 🔗
This is a complicated question and depends on several factors. A full regulatory strategy, as described in a previous question, would be able to provide a more nuanced answer.
You may want to check out our Ultimate 510(k) Checklist to see a very granular list of what goes into a 510(k).
Here are some general observations:
- Software teams that haven’t put together documentation for a 510(k) can take 6 - 12 months writing all of the documents needed.
- If you don’t plan ahead, clinical validation and performance testing can easily become bottle necks. In general, clinical validation and performance testing may take several months (or more) depending on the level of testing needed.
- Cybersecurity work, including both necessary software updates and documentation, can easily add months to the timeline.
- Putting together the bare minimum needed to submit is one thing, but if you miss key issues or documents, FDA will issue a hold letter and you’ll end up waiting anyway.
Once submitted, how long does it take to get clearance? 🔗
The FDA aims to make a decision within 90 days. However, if there are questions they need you to resolve, they’ll issue a hold letter by day 60. When FDA sends the request for additional information, their 90 day clock stops and your submission is placed on hold for up to 180 days. If you do not submit a formal response to FDA within the 180 days, FDA will delete your submission. The time it takes you to respond to a hold letter will further delay the submission. When you provide the response to the request for additional information, the 90 day FDA clock resumes. Also, while FDA tries to meet the 90 day review goal, they may not meet their goal.
You may want to look up how long it has taken for recent devices to be cleared within your device’s product code. This can give you some feeling for how long it may take you. You can use our FDA browser to look up this information. Keep in mind that even looking at recent devices isn’t always the best gauge of how long it will take, since it can also depend on the experience level of the other submitters.
Also, keep in mind that FDA reviews tend to slow down during the holidays.
How much does it cost to submit a 510(k)? 🔗
The biggest cost to submitting a 510(k) is preparing the software documentation and/or running the performance or clinical studies necessary for the submission. These can easily cost into the $100,000s.
The FDA also charges a few fees.
- Standard 510(k) Fee: $26,067
- Small Business 510(k) Fee: $6,517
- Annual Establishment Registration Fee: $11,423
These numbers are valid from October 1, 2025 through September 30, 2026. For the latest fees, please visit FDA’s Medical Device User Fee Amendments (MDUFA) website.
To get the Small Business 510(k) fee you’ll need to submit a Small Business Certification Request. Read here for more details about the process. We can also help with your small business application.
What is the difference between a Traditional, Special, and Abbreviated 510(k)? 🔗
There are three types of 510(k) submissions:
- Traditional
- Special
- Abbreviated
The Traditional 510(k) is almost always used for new SaMD devices. For modifications to previously cleared devices, the Special 510(k) is worth considering because it has a 30 day review goal rather than the 90 day goal for traditional and abbreviated 510(k)s.
The Abbreviated 510(k) pathway is less common. In 2022 there were only 48 Abbreviated 510(k)s that received clearance, as compared to 1000s of Traditional 510(k)s. Also, many of the Abbreviated 510(k)s were for latex gloves and other types of devices where there are clear performance standards that can be used for well-characterized devices. If your device is complex or relatively new, it’s unlikely an abbreviated 510(k) is appropriate. Furthermore, the FDA’s costs and review timelines are not shorter.
We sell our device in the EU already. Will that save us time on our 510(k)? 🔗
Yes. It will save a significant amount of time, since most of the software documentation you’ll have already. There are still significant differences and documentation between an EU Technical File and a 510(k) but it will get you about 60% of the way there and will save an equivalent amount of money.
De Novo Requests 🔗
What is a De Novo request? 🔗
A De Novo request is a request to classify a low to medium risk (class 1 or 2) medical device that doesn’t have any suitable predicate devices on the market.
A lot of the software documentation required for a De Novo is similar to a 510(k), however, there are also many differences. You likely will also need clinical evidence. Traditional 510(k)s often do not require clinical evidence.
One benefit of doing a De Novo is you can suggest the special controls for the new product code that is created, which can (in certain cases) act as a competitive moat.
See this section of the regulations for more details.
How common are De Novo classifications? 🔗
They’re much less common than 510(k)s.
How different is a De Novo compared to a 510(k)? 🔗
While much of the information is the same (administrative information, device description), De Novos require the following information which is not required for a 510(k):
- The classification being recommended under section 513 of the Federal Food, Drug, and Cosmetic Act (FD&C Act)
- A complete discussion of why general controls or general and special controls provide reasonable assurance of the safety and effectiveness of the device, and what special controls, if proposing a class II designation, would allow the Agency to conclude there is reasonable assurance the device is safe and effective for its intended use
- Clinical data (if applicable) that are relevant to support reasonable assurance of the safety and effectiveness of the device
- A description of the probable benefits of the device when compared to the probable or anticipated risks when the device is used as intended
How long does it take to put together a De Novo Classification Request? 🔗
Due to the additional information and likely need to perform clinical testing, it may easily take more than 12 months. Additionally, if you think that the De Novo pathway is applicable for your software device, we strongly suggest to have a pre-submission meeting with FDA.
Can Innolitics help with my De Novo request? 🔗
Yes! We have experience with De Novo submissions and would be happy to help.
What should we do if we’re on the edge between a 510(k) and De Novo? 🔗
In our experience, FDA really tries to push devices to follow the 510(k) pathway if there’s any possible way to do so. Thus, be aware if you submit a De Novo (which is more work), FDA may suggest a predicate device and push you over to being a 510(k).
On the flip side, if you do a 510(k) with a bad predicate, FDA may mark you as non-substantially equivalent.
In these cases, a 513(g) application and/or a pre-submission meeting would be recommended.
Clinical Investigations 🔗
What is a clinical investigation? 🔗
A clinical investigation is a broad term that refers to medical research studies involving people. It can have specific meanings in particular regulatory contexts. Generally it is used interchangeably with the term “clinical study”. Some medical devices require clinical studies to assess the performance of the device in a clinical setting.
Note that the term “clinical investigation” is the one that’s most often used in the regulations, however, because “clinical study” is shorter and more widely used, it is the term we typically use.
What is a clinical trial? 🔗
The terms clinical study, clinical investigation, and clinical trial are sometimes used interchangeably, but there’s a useful distinction to be made.
In most contexts, a clinical trial is a type of clinical study that
- involves human participants
- prospectively assigns participants to an intervention
- is designed to evaluate the effect of the intervention on the participants
- is evaluating a health-related biomedical or behavioral outcome.
Clinical trials are the primary way that researchers determine if a new form of treatment or prevention, such as a new drug, diet, or medical device (for example, a pacemaker), is safe and effective in people.
Unless exempted, clinical trials require the sponsor to submit an Investigational Device Exemption (IDE) application to FDA before initiating the trial. FDA will review the Institutional Review Board (IRB) approved study protocol and approve or deny the application.
Do all medical devices require a clinical studies or trials? 🔗
No. Most class 1 and many class 2 devices do not require clinical studies. According to FDA’s 2014 guidance, which is of course outdated, “FDA requests clinical data for less than 10 percent of 510(k) submissions.”
Some class 2 devices do require clinical studies. For example, Computer Aided Diagnostic devices, “CADe”, typically require a type of clinical investigation called a Multi-Reader Multi-Case (MRMC) study wherein readers try to diagnose a number of cases with and without the assistance of the CADe device being studied. This is not a clinical trial, but it is a clinical study.
Many digital therapeutic devices (see here for our article on this topic) require clinical trials.
It is likely that a De Novo request would include some degree of clinical information.
Non Compliance 🔗
How would FDA identify regulatory violations? 🔗
FDA has a few mechanisms of identifying regulatory violations.
If you are marketing a product that FDA believes is a medical device, yet you haven’t registered with FDA or completed the appropriate premarket submissions, then FDA will contact you. FDA may see marketing claims on your website, or other sales and marketing materials. Also, competitors can report allegations of regulatory misconduct on this website.
The form that FDA uses to contact you varies but may include informal phone calls, untitled letters and warning letters. Warning letters are placed on a public-facing FDA website and are sometimes used by your competition to dissuade potential customers from purchasing your device.
How often does FDA inspect medical device manufacturers? 🔗
Once you register with FDA and list your products they will periodically inspect you. Class 2 and 3 devices are inspected every two years or according to audit staff availability. FDA will also occasionally audit establishments who market class 1 devices (albeit less frequently than class 2 and 3 establishments).
What happens if FDA finds problems during the inspection? 🔗
FDA will issue an FDA Form 483 at the conclusion of an inspection if investigator(s) have observed anything they believe to be a violation. Often, the manufacturer is given an opportunity to correct the identified problems.
Depending on the severity and nature of the findings, FDA may also do any of the following:
- Issue a public warning letter
- Issue product recalls
- Seize products
- Issue court orders to stop certain activities
- Initiate criminal prosecutions
What happens if we sell a product but FDA believes it is a device? 🔗
Once it comes to FDA’s attention that you’re marketing a medical device without having followed the appropriate regulatory controls, such as a 510(k), FDA would notify you of the objectionable activities. They’d typically require that you stop marketing the device. If the device is already in use, you likely would need to issue a recall and pull the device off the market. If you are located outside of the US and attempting to import devices, then the product would likely be held up in customs. Finally, you’d have to comply with all of the regulatory controls for your device type before placing your device on the market again.
Clearly this would be a big problem, because you may lose most (or all) of your customers, your reputation would be ruined, and you’d have to complete a lot of regulatory work before you could resume marketing your device.
See the section below for examples.
What happens if we add new features to our 510(k) cleared software which change the intended use or classification but don’t submit a new 510(k)? 🔗
The next steps would be similar to the question above: FDA would notify you of the objectionable activities which may include pulling your device off the market (recall) or roll back the software version to the cleared version.
What if we make marketing or performance claims about our device which FDA believes are unsubstantiated? 🔗
In the case where you’re making marketing claims about your device which you can’t support with evidence, then FDA may notify you that you need to change the marketing material inline with your cleared intended use.
When developing a regulatory strategy, it’s important to consider the regulatory claims you’d like to be able to make. Often we suggest declaring the most important ones to FDA to de-risk their use and to ensure FDA agrees they’re substantiated by the evidence.
Regulatory Help 🔗
I’ve never submitted a 510(k) before. How do I get help? 🔗
You should probably hire a regulatory affairs consultant. Consultants can be expensive, but figuring out how to do everything on your own will likely cost even more, and mistakes can be time-consuming to correct.
What do regulatory affairs (RA) consultants do? 🔗
- Classification and Jurisdiction: Analyze a software function to determine if FDA, or other regulatory bodies, would consider it a medical device.
- Strategy: Put together a strategy for bringing a medical device onto the market in a way that maximally achieves the business goals (see previous questions about this).
- Gap Assessments: Compare devices and documentation against applicable regulations and standards to see if anything is missing.
- Prepare Submissions: Guide the team as they write documentation for regulatory submissions, and may write portions of the submissions.
- Regulatory Communication: Interfaces with regulatory agencies on your behalf.
- Quality Systems: Set up and maintain your quality system.
How do we find good regulatory consultants? 🔗
Innolitics is a great choice for Software as a Medical Device (SaMD) products. We offer several services, including our “Fast 510(k)” service for companies who have relatively little documentation, but nearly finished software and who are in a hurry. Our “Guided 510(k)” is a good fit for companies earlier in the process who would like guidance on regulatory strategy and putting together the submission.
Here are some other groups we can recommend:
- MDC Associates is a great group for In-Vitro Diagnostic Devices.
- MCRA is another group that has deep experience and many ex-FDA reviewers on their team.
- Medical Device Academy is an affordable group with fixed-fee pricing that has a focus on training.
- Lean RAQA is an affordable group who do great quality work.
You may also want to look through the FDA’s database to see who has submitted 510(k)s in your product code or similar product codes.
Is it a good idea to hire someone internally to handle both regulatory affairs and quality systems for your company? 🔗
- We recommend to distinguish between regulatory affairs and quality systems.
- Regulatory tends to be more involved early in the process as you classify your device, come up with a regulatory strategy, and then make your premarket submission (e.g., a 510(k) or a De Novo).
- Quality systems often starts coming into play a little later, but once your device is on the market, the volume of work for them tends to be steadier. As mentioned previously, we recommend starting work on your Quality Management System (QMS) after you have submitted your 510(k) and you are waiting for the review. In some cases, it makes sense to outsource most of your quality systems activities. We know a few groups we could recommend.
- Sometimes people try to combine the roles, however, we don’t suggest doing this as its often more expensive to find someone sufficiently qualified to handle both areas.
- Generally, once your device is on the market, the quality systems work tends to increase in volume more quickly than regulatory work, so we’ve found that it can make sense to either hire a full-time quality systems person or potentially outsource most of the quality work to a consultant.
Is it better to hire a full-time regulatory affairs person, or a consultant? 🔗
For most small or medium-sized companies who are making few regulatory submissions, it will be more cost effective to hire a consultant. As is always the case when working with consultants, there are downsides like limited availability, but it will be difficult and expensive to hire a full-time person with the level of experience you’ll want as a full-time staff.
Also, your needs for a regulatory consultant will vary throughout the project life-cycle. Typically, you will need help early on when developing a regulatory strategy. Then there will be a long lull as the development is completed. Then you will need help with performance studies, making the regulatory submission, and responding to hold letters. Thus, it can save significant costs by using a consultant whom you’re not paying through the long lull during development.
Is it feasible to develop a regulatory strategy by yourself? 🔗
We occasionally see early stage founders trying to develop a regulatory strategy on their own. We wouldn’t say it’s impossible, but it’s a risky approach. It’s easy to misinterpret the FDA guidance and regulations, and small misinterpretations can lead to large consequences. For example, if you believe you’re a class 1 device when you’re actually class 2, you’ll be in trouble when FDA comes knocking. Or, if you think your predicate can be device A, but FDA thinks it should be device B and furthermore you need additional clinical testing and evidence.
How do you pick a regulatory consultant to work with to help you submit a 510(k)? 🔗
You want a firm that has relevant and recent experience. In particular, you’d like a firm who has submitted a number of 510(k)s for devices somewhat similar to yours.
If your device is SaMD, then you also want to work with a firm that has successfully cleared 510(k)s for SaMDs. SaMD is very different than hardware-only devices and is also somewhat different from software and hardware devices too.
You’ll want to understand how busy the consultants are, and how many other clients they may be working with at the same time.
You’ll want to know if they’ll be able to help you to produce the documentation.
Why do different firms have such different prices? 🔗
The scope of service provided can vary a lot, and along with this variance, there is a large variance in cost. Thus, when comparing quotes from different firms, be sure you understand what is in scope or out of scope.
For example, we can just provide companies high-level guidance and templates, or we can help write most of the documentation for you. There is more than an order-of-magnitude difference between these services and their associated costs.
Engineering Help 🔗
Can we outsource our medical device software development? 🔗
Yes, although you—the medical-device manufacturer—are legally responsible for their work. You’ll still need a Quality Management System (QMS). As part of that QMS, you’ll need to have a purchasing process (section 820.50 of FDA Quality System Regulation, section 7.4.1, 7.4.2, and 7.4.3 of ISO 13485) where you keep records of the criteria to evaluate and select suppliers.
You’ll also still need to provide software validation documentation to FDA as part of your submission. Thus, you’ll want to be sure the software development team that produced the software will also be able to provide all of the necessary documents. Typically these details are arranged as part of a quality agreement.
What is a quality agreement? 🔗
A quality agreement is a signed document that states what parts of the Quality System regulation will be fulfilled by the software development firm (i.e., the supplier) and you the manufacturer.
How should we evaluate our software developers? 🔗
Since the software developers will need to provide documentation for your regulatory submissions, you’ll want to know they’ve done this before. You can ask them questions such as the following:
- What software documentation will you provide along with the source code? (Then cross check what they tell you with the 2023 software guidance.)
- Do you have a quality management system? Is it certified against ISO 13485?
- Do your software development processes comply with IEC 62304?
- Will you provide cybersecurity documentation? Does your security risk management comply with AAMI TIR 57 or ANSI/A AMI SW96:2023?
Is it reasonable to expect our software developers to provide the necessary regulatory documentation? 🔗
If it was part of what you agreed to up front, it would be reasonable. If you hired a software development firm that doesn’t work on medical devices, they won’t know what regulatory documentation they would be expected to provide. Not only that, it will be difficult for them to provide good documentation and it would be more than would typically be expected from an out-sourced development firm. For example, part of the software documentation is the software risk management assessment. This document requires input from clinical specialists who know how the device will be used in the clinic. A general purpose software engineering firm won’t be able to help with this.
Post Market 🔗
What do we have to do once we’ve gotten regulatory clearance? 🔗
NOTE: This response is incomplete and will be updated in a future revision.
Once you’ve gotten regulatory clearance for your SaMD product, you still need to:
- Obtain and maintain your annual Establishment Registration
- List your device(s)
- Implement and maintain your Quality Management System (QMS) processes, including:
- Design Control/Design History Files
- Management Review
- Device History Record
- Device Master Record
- Document Controls
- Purchasing Control
- Internal QMS Audit
- Collect Feedback from Customers
- Complaint Files
- Document, Investigate, and Report Safety Incidents
- Corrective and Preventative Action (CAPA)
- Document changes to your software and assess regulatory impact
- Be prepared for FDA Audits of your QMS
- Run Postmarket Cybersecurity Processes
- Periodic Penetration Tests
- Periodic Vulnerability Scans
- Assess, Remediate and Report Vulnerabilities
- Postmarket Surveillance
- Produce Unique Device Identifiers for New Versions
- Review marketing materials and Web site to assure alignment with clearance
What is Establishment Registration? 🔗
Companies that produce and sell medical devices in the U.S. must register annually with the FDA. This process is known as establishment registration. There is an annual fee. In 2024, it’s $7,653 for businesses of all sizes.
You can read more about the process on the FDA website.
How do Unique Device Identifiers work for SaMD? 🔗
FDA requires manufacturers to assign a unique device identifier (UDI) to each device. Each build of the software must have a new UDI. UDIs must be issued by an FDA-accredited issuing agency (i.e., GS1, HIBC). The UDI consists of two parts, a device identifier (DI) and a production identifier (PI). This applies to both hardware and software devices. Please see the GS1 example below for an explanation.
(01)506060888065(8018)1.0.0(11)2024-01-25
The #s in parentheses are called Application Identifiers (AIs)and are assigned meanings. In this example (01) designates the Device Identifier (DI). This is created using GS1 software. (8018) designates the software version and (11) designates the date of manufacture (software release date). The Production Identifier is the software version (1.0.0) and the release date (2024-01-25)
The UDI can be either be a barcode with human readable characters below the barcode or can be the human readable characters without the barcode.
Once you have the UDI(s), you will need to create an account to FDA’s Global Unique Identification Database (GUDID). You will enter the UDI information in the database. There is no fee for this.
For a software medical device (SaMD) that has a user interface, it’s typical to show the UDI as a part of the device label in an about page or help box. The device label (with UDI) must not be on a transient screen like the software startup screen. For SaMD that does not have a user interface (e.g., software that automatically processes data and produces a report), we typically show the UDI somewhere in the generated reports.
The only external cost will be for the subscription to the Accredited Issuing Agency (i.e., GS1). If you only have one device the current GS1 initial fee is $30 USD. The initial fee for up to 10 UDI’s is $250 with a $50 annual fee. The other Accredited Issuing Agencies fees should be comparable. You should be able to create each UDI and enter it in the GUDID database in just a few hours.
How does Medical Device Tracking work for SaMD? 🔗
We’re not aware of any SaMD that must comply with the Medical Device Tracking regulations.
Medical device tracking is meant to make it easier to inform patients and clinicians when there is a device recall. Only certain types of devices are tracked—these are referred to as “tracked devices” in 21 CFR 821. In FDA’s 2014 guidance Medical Device Tracking, they list the product codes which require tracking. Most of them are implanted devices (e.g., pacemakers, heart valves, or prosthesis) or devices that sustain patient health (e.g., infusion pumps, ventilators, or breathing monitors). If your device does require medical device tracking, FDA will notify you of this when you receive clearance or approval to market your device.
Once we get our software cleared by the FDA, can we keep updating it? 🔗
Yes, you can keep updating the software. In fact, FDA expects that you will continue providing cybersecurity updates. That said, if the changes to the software are significant, you may need to submit a new 510(k), including possibly collecting new clinical or performance data.
Here is the flow chart from FDA’s 2017 Guidance, Deciding When to Submit a 510(k) for a Software Change to an Existing Device:
- Where research ends
- After feasibility / proof of concept
- Pre-market
- Prior to start of investigation device exemption
- Mechanism of change/revision
In addition to the software change guidance you will also need to work through the various flowcharts in FDA’s general change guidance.
There is some strategy surrounding the order that features are developed. For example, it is often a good idea to create key features that FDA needs to evaluate in v1.0, but then to have the software engineering team work on the v1.1 features while you’re waiting for FDA to review your submission.
Checkout our article, How Frequently Can you Release Medical Device Software for additional information on this topic.
Examples 🔗
Software-Related FDA Product Codes 🔗
You can see all of the product-codes in the FDA’s Product Classification database or our FDA Browser tool (which is a user-friendly portal that sync’s with FDA’s database.)
Here is a list of product codes:
| Panel | Regulation | Product Code | Name | Class |
|---|---|---|---|---|
| Clinical Chemistry | 862.1055 | NQL | System, Test, Amino Acids, Free Carnitines And Acylcarnitines Tandem Mass Spectrometry | 2 |
| Clinical Chemistry | 862.1356 | QJI | Interoperable Automated Glycemic Controller | 2 |
| Clinical Chemistry | 862.1356 | QJS | Interoperable Automated Glycemic Controller, Insulin Suspend | 2 |
| Toxicology | 862.2265 | PFF | High Throughput Dna Sequence Analyzer | 2 |
| Radiology | 892.2050 | LLZ | System, Image Processing, Radiological | 2 |
| Ophthalmic | 892.2050 | NFJ | System, Image Management, Ophthalmic | 2 |
| Radiology | 892.2050 | NWE | Colon Computed Tomography System, Computer Aided Detection | 2 |
| Radiology | 892.2050 | OEB | Lung Computed Tomography System, Computer-Aided Detection | 2 |
| Radiology | 892.2050 | OMJ | Chest X-Ray Computer Aided Detection | 2 |
| Radiology | 892.2050 | PGY | Display, Diagnostic Radiology | 2 |
| Cardiovascular | 892.2050 | PZO | Software For Visualization Of Vascular Anatomy And Intravascular Devices | 2 |
| Radiology | 892.2050 | QIH | Automated Radiological Image Processing Software | 2 |
| Radiology | 892.2050 | QKB | Radiological Image Processing Software For Radiation Therapy | 2 |
| Ear Nose & Throat | 892.2050 | QQE | Image Management Software For Planning Of Otologic And Neurotologic Procedures | 2 |
| Radiology | 892.2050 | QTZ | Radiological Image Processing Software For Ablation Therapy Planning And Evaluation | 2 |
| 892.2265 | PFF | High Throughput DNA Sequence Analyzer | 2 | |
| Pathology | 864.3750 | QPN | Software Algorithm Device To Assist Users In Digital Pathology | 2 |
| Pathology | 864.3700 | PSY | Whole Slide Imaging System | 2 |
| Pathology | 864.3700 | PZZ | Digital Pathology Display | 2 |
| Pathology | 864.3700 | QKQ | Digital Pathology Image Viewing And Management Software | 2 |
| Hematology | 864.9165 | MMH | Blood Establishment Computer Software And Accessories | 2 |
| Microbiology | 866.2190 | PPU | Microbial Colony Image Assessment System | 2 |
| Microbiology | 866.2190 | QQY | Culture Plate Imaging System For Qualitative Assessment Of Resistant Organisms | 2 |
| Cardiovascular | 870.1405 | QQI | Interventional Cardiovascular Implant Simulation Software Device | 2 |
| Cardiovascular | 870.1415 | PJA | Coronary Vascular Physiologic Simulation Software | 2 |
| Cardiovascular | 870.1415 | QEK | Angiographic Coronary Vascular Physiologic Simulation Software | 2 |
| Dental | 872.3661 | KZN | Scanner, Color | 2 |
| Dental | 872.3661 | NOF | System, Optical Impression, Computer Assisted Design And Manufacturing (Cad/Cam) Of Dental Restorations | 2 |
| Dental | 872.3661 | QJK | Intraoral Camera | 2 |
| Ear Nose & Throat | 874.3315 | PLK | Tympanic Membrane Direct Contact Hearing Aid | 2 |
| Ear Nose & Throat | 874.3325 | QDD | Self-Fitting Air-Conduction Hearing Aid, Prescription | 2 |
| Ear Nose & Throat | 874.3325 | QUH | Self-Fitting Air-Conduction Hearing Aid, Over The Counter | 2 |
| Ear Nose & Throat | 874.3340 | PFO | Active Implantable Bone Conduction Hearing System | 2 |
| Gastroenterology/Urology | 876.1300 | NEZ | System, Imaging, Gastrointestinal, Wireless, Capsule | 2 |
| Gastroenterology/Urology | 876.1300 | NSI | System, Imaging, Esophageal, Wireless, Capsule | 2 |
| Gastroenterology/Urology | 876.1310 | QKZ | Magnetically Maneuvered Capsule Endoscopy System | 2 |
| Gastroenterology/Urology | 876.1330 | PGD | Colon Capsule Imaging System | 2 |
| Gastroenterology/Urology | 876.1450 | QIS | Esophageal, Mucosal, Electrical Characterization | 2 |
| Gastroenterology/Urology | 876.1520 | QNP | Gastrointesinal Lesion Software Detection System | 2 |
| Gastroenterology/Urology | 876.1735 | MYE | System, Electrogastrography (Egg) | 2 |
| Gastroenterology/Urology | 876.2050 | OQT | Prostate Lesion, Documentation, System | 2 |
| Gastroenterology/Urology | 876.4340 | PLP | High Intensity Ultrasound System For Prostate Tissue Ablation | 2 |
| Gastroenterology/Urology | 876.4350 | PZP | Fluid Jet Removal System | 2 |
| Gastroenterology/Urology | 876.5026 | QRC | Non-Implanted Electrical Stimulation Device For Management Of Premature Ejaculation | 2 |
| Gastroenterology/Urology | 876.5330 | QAJ | Cutaneous Electrode Stimulator For Urinary Incontinence | 2 |
| Neurology | 876.5340 | QHH | Non-Implanted Nerve Stimulator For Pain Associated With Irritable Bowel Syndrome (Ibs) | 2 |
| Gastroenterology/Urology | 876.5960 | QMY | Computerized Behavioral Therapy Device For Treating Symptoms | 2 |
| Gastroenterology/Urology | 876.5981 | ONY | Oral Removable Retainer For Weight Management | 2 |
| General & Plastic Surgery | 878.3510 | PQN | Carbon Dioxide Gas Controlled Tissue Expander | 2 |
| General & Plastic Surgery | 878.4360 | PMC | Scalp Cooling System | 2 |
| General & Plastic Surgery | 878.4420 | PAY | Over-The-Counter Radiofrequency Coagulation Device For Wrinkle Reduction | 2 |
| General & Plastic Surgery | 878.4430 | QAI | Powered Microneedle Device | 2 |
| General & Plastic Surgery | 878.4550 | QDF | Parathyroid Autofluorescence Detection Device | 2 |
| General & Plastic Surgery | 878.4550 | QDG | Parathyroid Autofluorescence Imaging Device | 2 |
| General & Plastic Surgery | 878.4550 | QJF | Autofluorescence Imaging Adjunct Tool For Wounds | 2 |
| General & Plastic Surgery | 878.4635 | LEJ | Booth, Sun Tan | 2 |
| General & Plastic Surgery | 878.4635 | RAB | Sunlamp Products (Pre-Standard) | 2 |
| General & Plastic Surgery | 878.4635 | REF | Suntan Bed | 2 |
| General & Plastic Surgery | 878.4635 | REG | Suntan Lamp | 2 |
| General & Plastic Surgery | 878.4635 | REH | Tabletop Sunlamp System | 2 |
| General & Plastic Surgery | 878.4685 | PZL | Extracorporeal Shock Wave Device For Treatment Of Diabetic Foot Ulcers | 2 |
| General & Plastic Surgery | 878.4740 | GAG | Stapler, Surgical | 2 |
| General & Plastic Surgery | 878.4740 | QQS | Stapler, Skin | 1 |
| General & Plastic Surgery | 878.4783 | QFC | Negative Pressure Wound Therapy Device For Reduction Of Wound Complications | 2 |
| General & Plastic Surgery | 878.4961 | QNM | Mountable Electromechanical Surgical System For Transluminal Approaches | 2 |
| General Hospital | 878.5050 | PQM | Surgical Smoke Precipitator | 2 |
| General & Plastic Surgery | 880.2750 | PBZ | Image Processing Device For Estimation Of External Blood Loss | 2 |
| General Hospital | 880.5445 | OKL | Intravascular Administration Set, Automated Air Removal System | 2 |
| General Hospital | 880.6210 | MTV | Sharps Needle Destruction Device | 2 |
| General Hospital | 880.6315 | NZH | Medication Management System, Remote | 2 |
| General Hospital | 880.6600 | OSZ | Antimicrobial Keyboard | 2 |
| Neurology | 882.1440 | NCG | Neuropsychiatric Interpretative Electroencephalograph Assessment Aid | 2 |
| Neurology | 882.1450 | PIW | Brain Injury Adjunctive Interpretive Electroencephalograph Assessment Aid | 2 |
| Neurology | 882.1455 | QEA | Brain Injury Adjunctive Interpretive Oculomotor Assessment Aid | 2 |
| Neurology | 882.1470 | PKQ | Computerized Cognitive Assessment Aid | 2 |
| Neurologyw | 882.1470 | PTY | Computerized Cognitive Assessment Aid, Exempt | 2 |
| Neurology | 882.1471 | POM | Computerized Cognitive Assessment Aid For Concussion | 2 |
| Neurology | 882.1491 | QPF | Pediatric Autism Spectrum Disorder Diagnosis Aid | 2 |
| Neurology | 882.1580 | POS | Physiological Signal Based Seizure Monitoring System | 2 |
| Neurology | 882.1630 | POP | Cranial Motion Measurement Device | 2 |
| Neurology | 882.1935 | OPT | Infrared Hematoma Detector | 2 |
| 882.5056 | ||||
| Neurology | 882.5700 | PLU | Thermal System For Insomnia | 2 |
| Neurology | 882.5705 | QMZ | Digital Therapy Device To Reduce Sleep Disturbance For Psychiatric Conditions | 2 |
| Neurology | 882.5800 | JXK | Cranial Electrotherapy Stimulator To Treat Depression | 3 |
| Neurology | 882.5800 | QJQ | Cranial Electrotherapy Stimulator To Treat Insomnia And/Or Anxiety | 2 |
| Neurology | 882.5801 | PWE | Computerized Behavioral Therapy Device For Substance Use Disorders | 2 |
| Neurology | 882.5801 | QVO | Computerized Behavioral Therapy Device For Insomnia | 2 |
| Neurology | 882.5802 | QCI | Transcranial Magnetic Stimulation System For Obsessive-Compulsive Disorder | 2 |
| Neurology | 882.5802 | QMD | Transcranial Magnetic Stimulation System For Smoking Cessation | 2 |
| Neurology | 882.5808 | OKP | Transcranial Magnetic Stimulator For The Treatment Of Migraine Headache | 2 |
| Neurology | 882.5855 | QQC | Brain Stimulation Programming Planning Software. | 2 |
| Neurology | 882.5891 | PCC | Stimulator, Nerve, Electrical, Transcutaneous, For Migraine | 2 |
| Neurology | 882.5892 | PKR | Non-Invasive Vagus Nerve Stimulator - Headache | 2 |
| Neurology | 882.5892 | QAK | Non-Invasive Vagus Nerve Stimulator For Migraine Headache | 2 |
| Neurology | 882.5893 | QAR | Thermal Vestibular Stimulator For Headache | 2 |
| Neurology | 882.5895 | OVP | Vibratory Counter-Stimulation | 2 |
| Neurology | 882.5896 | PZR | Percutaneous Nerve Stimulator For Opioid Withdrawal | 2 |
| Neurology | 882.5897 | QBC | External Upper Limb Tremor Stimulator | 2 |
| Neurology | 882.5898 | QGL | Transcutaneous Nerve Stimulator For Adhd | 2 |
| Neurology | 882.5940 | GXC | Device, Electroconvulsive Therapy | 3 |
| Neurology | 882.5940 | QGH | Electroconvulsive Therapy Device For Catatonia, Major Depressive Disorder, And Bipolar Disorder | 2 |
| Obstetrics/Gynecology | 884.1630 | HEX | Colposcope (And Colpomicroscope) | 2 |
| Obstetrics/Gynecology | 884.1630 | MOK | Vaginoscope And Accessories | 2 |
| Obstetrics/Gynecology | 884.1630 | OHE | Forensic Evidence Sexual Assault Kit | 2 |
| Obstetrics/Gynecology | 884.1630 | PTZ | Colposcope (And Colpomicroscope), Exempt | 2 |
| Obstetrics/Gynecology | 884.1710 | PGT | Insufflator, Hysteroscopic, Fluid, Closed-Loop Recirculation With Cutter-Coagulator, Endoscopic, Bipolar | 2 |
| Obstetrics/Gynecology | 884.5370 | PYT | Device, Fertility Diagnostic, Contraceptive, Software Application | 2 |
| Obstetrics/Gynecology | 884.6195 | PBH | Embryo Image Assessment System, Assisted Reproduction | 2 |
| Ophthalmic | 886.1100 | PIB | Diabetic Retinopathy Detection Device | 2 |
| Ophthalmic | 886.1342 | PMW | Strabismus Detection Device | 2 |
| Ophthalmic | 886.1450 | HJB | Instrument, Measuring, Corneal Radius | 1 |
| Ophthalmic | 886.1925 | PLZ | Ocular Pattern Recorder | 2 |
| Ophthalmic | 886.5201 | QIU | Intense Pulsed Light Device For Managing Dry Eye | 2 |
| Ophthalmic | 886.5300 | PQJ | Intranasal Electrostimulation Device | 2 |
| Ophthalmic | 886.5305 | QKV | Electromechanical Tear Stimulator | 2 |
| Ophthalmic | 886.5310 | QBR | Intranasal Electrostimulation Device For Dry Eye Symptoms | 2 |
| Ophthalmic | 886.5905 | PIC | Oral Electronic Vision Aid | 2 |
| Orthopedic | 888.1600 | QGQ | Bone Indentation Device | 2 |
| Orthopedic | 888.3090 | QFP | Intraoperative Orthopedic Strain Sensor | 2 |
| Orthopedic | 888.3600 | QPP | Implantable Post-Surgical Kinematic Measurement Knee Device | 2 |
| Physical Medicine | 890.3110 | INO | Chair, Positioning, Electric | 2 |
| Physical Medicine | 890.3450 | PAE | Upper Extremity Prosthesis With Multiple Simultaneous Degrees Of Freedom And Controlled Via Cutaneous Electromyography | 2 |
| Neurology | 890.3480 | PHL | Powered Exoskeleton | 2 |
| Physical Medicine | 890.3690 | INK | Stretcher, Wheeled, Powered | 2 |
| Physical Medicine | 890.3890 | IMK | Wheelchair, Stair Climbing | 2 |
| Physical Medicine | 890.5420 | QOL | Electroencephalography (Eeg)-Driven Upper Extremity Powered Exerciser | 2 |
| Physical Medicine | 890.5525 | EGJ | Device, Iontophoresis, Other Uses | 2 |
| Physical Medicine | 890.5525 | KTB | Device, Iontophoresis, Specific Uses | 2 |
| Physical Medicine | 890.5670 | OSD | Massager, Therapeutic, To Internally Massage Trigger Points In The Pelvic Floor Musculature | 2 |
| Physical Medicine | 890.5670 | QKD | Massager, Therapeutic, To Internally Massage Trigger Points In The Pelvic Floor Musculature, Prescription Use | 2 |
| Physical Medicine | 890.5800 | QRA | Virtual Reality Behavioral Therapy Device For Pain Relief | 2 |
Special Controls 🔗
QME - Software For Optical Camera-Based Measurement Of Pulse Rate, Heart Rate, etc. 🔗
The QME product code (“Software For Optical Camera-Based Measurement Of Pulse Rate, Heart Rate, Breathing Rate, And/Or Respiratory Rate”) is Class 2. Manufacturers of these devices thus must comply with general controls and also special controls. The regulation number for the QME product code is 21 CFR 870.2785. All of this can be seen with Innolitics’ FDA browser or in the FDA’s product code database. Within the regulation, it states that the following special controls apply:
Classification. Class II (special controls). The special controls for this device are:
- A software description and the results of verification and validation testing based on a comprehensive hazard analysis and risk assessment must include:
- A full characterization of the software technical parameters, including algorithms;
- If required image acquisition hardware is not included with the device, full specifications of the hardware requirements and testing to demonstrate the specified hardware ensures adequate data for validated and accurate measurements;
- A description of the expected impact of all applicable sensor acquisition hardware characteristics and associated hardware specifications;
- A description of all mitigations for user error or failure of any subsystem components (including signal detection, signal analysis, data display, and storage) on output accuracy; and
- Software documentation must include a cybersecurity vulnerability and management process to assure software functionality.
- Clinical data must be provided. This assessment must fulfill the following:
- The clinical data must be representative of the intended use population for the device. Any selection criteria or sample limitations must be fully described and justified.
- The assessment must demonstrate output consistency using the expected range of data sources and data quality encountered in the intended use population and environment.
- The assessment must compare device output with a clinically accurate patient-contacting relevant comparator device in an accurate and reproducible manner.
- A human factors and usability engineering assessment must be provided that evaluates the risk of improper measurement.
- Labeling must include:
- A description of what the device measures and outputs to the user;
- Warnings identifying sensor acquisition factors or subject conditions or characteristics (garment types/textures, motion, etc.) that may impact measurement results;
- Guidance for interpretation of the measurements, including a statement that the output is adjunctive to other physical vital sign parameters and patient information;
- The expected performance of the device for all intended use populations and environments; and
- Robust instructions to ensure correct system setup.
QPN - Software Algorithm Device To Assist Users In Digital Pathology 🔗
The QPN product code (“Software Algorithm Device To Assist Users In Digital Pathology”) is Class 2. Manufacturers of these devices thus must comply with general controls and also special controls. The regulation number for the QPN product code is 21 CFR 864.3750. This product code was created in 2020 due to a De Novo request for the Paige Prostate software (here’s their Decision summary). Within the regulation, it states that the following description of the type of device:
A software algorithm device to assist users in digital pathology is an in vitro diagnostic device intended to evaluate acquired scanned pathology whole slide images. The device uses software algorithms to provide information to the user about presence, location, and characteristics of areas of the image with clinical implications. Information from this device is intended to assist the user in determining a pathology diagnosis.
It then states that the following special controls apply:
Classification. Class II (special controls). The special controls for this device are:
- The intended use on the device's label and labeling required under § 809.10 of this chapter must include:
- Specimen type;
- Information on the device input(s) (e.g., scanned whole slide images (WSI), etc.);
- Information on the device output(s) (e.g., format of the information provided by the device to the user that can be used to evaluate the WSI, etc.);
- Intended users;
- Necessary input/output devices (e.g., WSI scanners, viewing software, etc.);
- A limiting statement that addresses use of the device as an adjunct; and
- A limiting statement that users should use the device in conjunction with complete standard of care evaluation of the WSI.
- The labeling required under § 809.10(b) of this chapter must include:
- A detailed description of the device, including the following:
- Detailed descriptions of the software device, including the detection/analysis algorithm, software design architecture, interaction with input/output devices, and necessary third-party software;
- Detailed descriptions of the intended user(s) and recommended training for safe use of the device; and
- Clear instructions about how to resolve device-related issues (e.g., cybersecurity or device malfunction issues).
- A detailed summary of the performance testing, including test methods, dataset characteristics, results, and a summary of sub-analyses on case distributions stratified by relevant confounders, such as anatomical characteristics, patient demographics, medical history, user experience, and scanning equipment, as applicable.
- Limiting statements that indicate:
- A description of situations in which the device may fail or may not operate at its expected performance level (e.g., poor image quality or for certain subpopulations), including any limitations in the dataset used to train, test, and tune the algorithm during device development;
- The data acquired using the device should only be interpreted by the types of users indicated in the intended use statement; and
- Qualified users should employ appropriate procedures and safeguards (e.g., quality control measures, etc.) to assure the validity of the interpretation of images obtained using this device.
- Design verification and validation must include:
- A detailed description of the device software, including its algorithm and its development, that includes a description of any datasets used to train, tune, or test the software algorithm. This detailed description of the device software must include:
- A detailed description of the technical performance assessment study protocols (e.g., regions of interest (ROI) localization study) and results used to assess the device output(s) (e.g., image overlays, image heatmaps, etc.);
- The training dataset must include cases representing different pre-analytical variables representative of the conditions likely to be encountered when used as intended (e.g., fixation type and time, histology slide processing techniques, challenging diagnostic cases, multiple sites, patient demographics, etc.);
- The number of WSI in an independent validation dataset must be appropriate to demonstrate device accuracy in detecting and localizing ROIs on scanned WSI, and must include subsets clinically relevant to the intended use of the device;
- Emergency recovery/backup functions, which must be included in the device design;
- System level architecture diagram with a matrix to depict the communication endpoints, communication protocols, and security protections for the device and its supportive systems, including any products or services that are included in the communication pathway; and
- A risk management plan, including a justification of how the cybersecurity vulnerabilities of third-party software and services are reduced by the device's risk management mitigations in order to address cybersecurity risks associated with key device functionality (such as loss of image, altered metadata, corrupted image data, degraded image quality, etc.). The risk management plan must also include how the device will be maintained on its intended platform (e.g. a general purpose computing platform, virtual machine, middleware, cloud-based computing services, medical device hardware, etc.), which includes how the software integrity will be maintained, how the software will be authenticated on the platform, how any reliance on the platform will be managed in order to facilitate implementation of cybersecurity controls (such as user authentication, communication encryption and authentication, etc.), and how the device will be protected when the underlying platform is not updated, such that the specific risks of the device are addressed (such as loss of image, altered metadata, corrupted image data, degraded image quality, etc.).
- Data demonstrating acceptable, as determined by FDA, analytical device performance, by conducting analytical studies. For each analytical study, relevant details must be documented (e.g., the origin of the study slides and images, reader/annotator qualifications, method of annotation, location of the study site(s), challenging diagnoses, etc.). The analytical studies must include:
- Bench testing or technical testing to assess device output, such as localization of ROIs within a pre-specified threshold. Samples must be representative of the entire spectrum of challenging cases likely to be encountered when the device is used as intended; and
- Data from a precision study that demonstrates device performance when used with multiple input devices (e.g., WSI scanners) to assess total variability across operators, within-scanner, between-scanner and between-site, using clinical specimens with defined, clinically relevant, and challenging characteristics likely to be encountered when the device is used as intended. Samples must be representative of the entire spectrum of challenging cases likely to be encountered when the device is used as intended. Precision, including performance of the device and reproducibility, must be assessed by agreement between replicates.
- Data demonstrating acceptable, as determined by FDA, clinical validation must be demonstrated by conducting studies with clinical specimens. For each clinical study, relevant details must be documented (e.g., the origin of the study slides and images, reader/annotator qualifications, method of annotation, location of the study site(s) (on-site/remote), challenging diagnoses, etc.). The studies must include:
- A study demonstrating the performance by the intended users with and without the software device (e.g., unassisted and device-assisted reading of scanned WSI of pathology slides). The study dataset must contain sufficient numbers of cases from relevant cohorts that are representative of the scope of patients likely to be encountered given the intended use of the device (e.g., subsets defined by clinically relevant confounders, challenging diagnoses, subsets with potential biopsy appearance modifiers, concomitant diseases, and subsets defined by image scanning characteristics, etc.) such that the performance estimates and confidence intervals for these individual subsets can be characterized. The performance assessment must be based on appropriate diagnostic accuracy measures (e.g., sensitivity, specificity, predictive value, diagnostic likelihood ratio, etc.).
- [Reserved]
- A detailed description of the device software, including its algorithm and its development, that includes a description of any datasets used to train, tune, or test the software algorithm. This detailed description of the device software must include:
- A detailed description of the device, including the following:
When reviewing the Paige Prostate Decision summary, you can see the Intended Use follows the special controls closely:
Paige Prostate is a software only device intended to assist pathologists in the detection of foci that are suspicious for cancer during the review of scanned whole slide images (WSI) from prostate needle biopsies prepared from hematoxylin & eosin (H&E) stained formalin-fixed paraffin embedded (FFPE) tissue. After initial diagnostic review of the WSI by the pathologist, if Paige Prostate detects tissue morphology suspicious for cancer, it provides coordinates (X,Y) on a single location on the image with the highest likelihood of having cancer for further review by the pathologist.
Paige Prostate is intended to be used with slide images digitized with Philips Ultra Fast
Scanner and visualized with Paige FullFocus WSI viewing software.
Paige Prostate is an adjunctive computer-assisted methodology and its output should not be used as the primary diagnosis. Pathologists should only use Paige Prostate in conjunction with their complete standard of care evaluation of the slide image.
Note that the special controls require a clinical study.
Warning Letters 🔗
Owlet Baby Care - Smart Sock Plus 🔗
This example shows what happens if a company markets (i.e., advertises a product for sale) that FDA believes is a medical device, without complying with the regulations.
Owlet sold the “Owlet Smart Sock” product, and made marketing claims such as:
- “Track the most important indicators of your baby’s health–like oxygen level, heart rate, and total hours slept”
- “If your baby’s readings leave preset “safe” zones, the Smart Sock will immediately notify you that your baby needs your attention.”
- “Rest easy knowing you’ll receive proactive notifications via lights and sounds if your baby’s oxygen level or heart rate leave preset zones”
FDA in their Warning Letter CMS 616354 on October 5, 2021, stated the following:
The United States Food and Drug Administration (FDA) has learned that your firm is marketing Owlet Smart Socks in the United States without marketing clearance or approval, in violation of the Federal Food, Drug, and Cosmetic Act (the Act) … these products are devices because they are intended for use in the diagnosis of disease or other conditions or in the cure, mitigation, treatment, or prevention of disease, or to affect the structure or any function of the body. Products that measure blood oxygen saturation and pulse rate are devices when they are intended to identify (diagnose) desaturation and bradycardia and provide an alarm to notify users that measurements are outside preset values.
Our office requests that Owlet Baby Care, Inc. cease any activities that result in the adulteration of the Owlet Smart Sock (All versions and co-branded products), such as the commercial distribution of the device for the uses discussed above.
In May 25th, 2023, Owlet was able to get 510(k) clearance for the following intended use:
The BabySat 3 pulse oximeter is indicated for use in measuring and displaying functional oxygen saturation of arterial hemoglobin (SpO2) and Pulse rate. It is indicated for spot-checking and/or continuous monitoring of well-perfused patients greater than one month old up to 18 months old and weighing between 6 and 30 lbs., in the home environment.
As part of their 510(k), they completed:
- Electrical Safety, EMC, RFID, and Wireless Co-existing testing per various international standards
- Software verification and validation
- Desaturation studies across the full Fitzpatrick Scale score
- Signal Quality Studies and At Home Studies
- Observational studies for skin irritation and signal quality data
- Human factors and usability studies.
As you can imagine, this was a lot of work.
You can read the full 510(k) summary letter online.
Revision History 🔗
| Date | Summary of Change |
|---|---|
| 8 December 2023 | Initial Version; It’s still rough around the edges, but useful enough to publish. |
| 6 January 2023 | Extend the section about post-market activities. Add more details about working with software engineering groups. |
| 29 January 2023 | Jim Luker, our VP of Regulatory Affairs, did a thorough review of the document and made several clarifications and a few small corrections. |


