Summary, Audience, Purpose 🔗
This article is a distillation of the 27 page FDA guidance document ‘Multiple Function Device Products: Policy and Considerations’. The audience is mainly regulatory affairs specialists and managers. Its purpose is to provide the core information from the guidance in a readable format.
Background 🔗
On December 13, 2016, a law called the Cures Act was signed, which clarified rules about medical software. One part of this law, called Section 3060(a), made changes to the FD&C Act, a set of rules that regulate food, drugs, and medical devices. These changes introduced a new section, 520(o), which explains what types of software are not considered medical devices and are therefore not regulated under the same rules.
The new section, 520(o)(2), talks about software products that have multiple functions, including at least one medical device function and at least one non-device software function. According to this section, the government will not regulate the non-device software part of these products as if they were medical devices. However, when the government checks if the medical device part is safe and effective, they can also look at how the non-device software part might affect the medical device part.
Scope 🔗
This article explains FDA’s regulatory approach and policy for multiple function device products. It is based on FDA’s 2020 Multiple Function Device Products: Policy and Considerations guidance.
Introduction 🔗
Multiple Function Device Products (MFDP) are products which include at least one medical device function and at least one non-device software function (other function).
Let's break down some important terms related to these products:
- Function: A distinct purpose of a product. For example, a product can have three functions: storage, transfer, and analysis.
- Medical Device function: A function that is considered a medical device under section 201(h) of the FD&C Act.
- Other function:
- A function that is not a medical device,
- is a medical device but not subject to premarket review, or
- is a medical device for which the FDA doesn't enforce compliance with regulatory controls.
- Device function-under-review: A function that the FDA is reviewing before the product goes to market.
When the FDA reviews MFDPs, they focus on the medical device function-under-review, which requires premarket clearance or approval. The same principles apply to all MFDPs, whether they are software-based, hardware-based, or both. This article will focus on software medical devices (SaMDs)
Assessing the Impact of Other Functions on Multiple Function Device Products 🔗
Manufacturers should determine and document if an "other function" affects the safety or effectiveness of the device function-under-review in their risk assessment process. These impacts can be negative or positive. For MFDPs, manufacturers should include information about the impacts of "other functions" in their premarket submissions only if they could negatively impact the device function-under-review or if they have a positive impact that will be mentioned in the device's labeling.
Although the FDA doesn't recommend submitting information about all positive impacts from "other functions" in a premarket submission, manufacturers must perform and document all impact assessments (negative, positive, and no impact) as part of the design validation process. These assessments are subject to FDA review during inspections.
Considerations for Multiple Function Device Products 🔗
The following aspects should be taken into consideration for MFDPs.
Designing and Implementing Separation 🔗
When designing and implementing a multiple function device product, it's important to separate the device function-under-review from the "other functions" as much as possible. This can be done through:
- logical separation,
- architectural separation,
- code, and data partitioning.
Using separation and partitions in software systems can help prevent "other functions" from negatively impacting the device function-under-review and reduce the risk of hazardous situations.
Documenting the results of a thorough risk analysis of the impacts of "other functions" and the mitigation strategies employed is crucial for an effective risk management process. The more separation there is between the functions, the easier it is to independently review the safety and effectiveness of the device function-under-review. Separation also increases the likelihood that the device function-under-review isn't dependent on the "other function(s)" in the product.
Making architecture decisions early in the design cycle can help achieve optimal separation and support the segregation needed for risk control. If separation isn't possible, the interconnection and interdependencies between the device function-under-review and "other functions" should be explained and included in the hazard analysis. Appropriate controls should be created to reduce any adverse impact of the connectivity on the safety and effectiveness of the device function-under-review.
When it comes to cybersecurity risks, some level of separation between the device function-under-review and the "other function(s)" may be necessary to mitigate those risks. For instance, modular or separation architectures can help reduce the possibility that a cybersecurity threat to or from an "other function" could impact the device function-under-review.
Factors to Consider for Other Functions 🔗
Manufacturers of MFDPs should take into account the following factors regarding all "other functions" of the product:
- Role of the Other Function: Consider how the "other function" affects the performance of the device function-under-review.
- Limitations: Identify any limitations of the device function-under-review when used in conjunction with the "other function."
- Resource Specifications: Develop appropriate hardware and software resource specifications for the product with multiple functions to minimize the impact of the "other function" on the performance of the device function-under-review.
- Ensuring Appropriate Actions: Make sure that end-users take appropriate actions when using the device function-under-review alongside the "other function."
- Risk Identification and Mitigation: Identify, evaluate, and mitigate any additional risks, including cybersecurity risks, in the device function-under-review when used in combination with the "other function."
By addressing these factors, manufacturers can better understand the potential impact of "other functions" on the device function-under-review and take necessary steps to ensure the safety and effectiveness of their product.
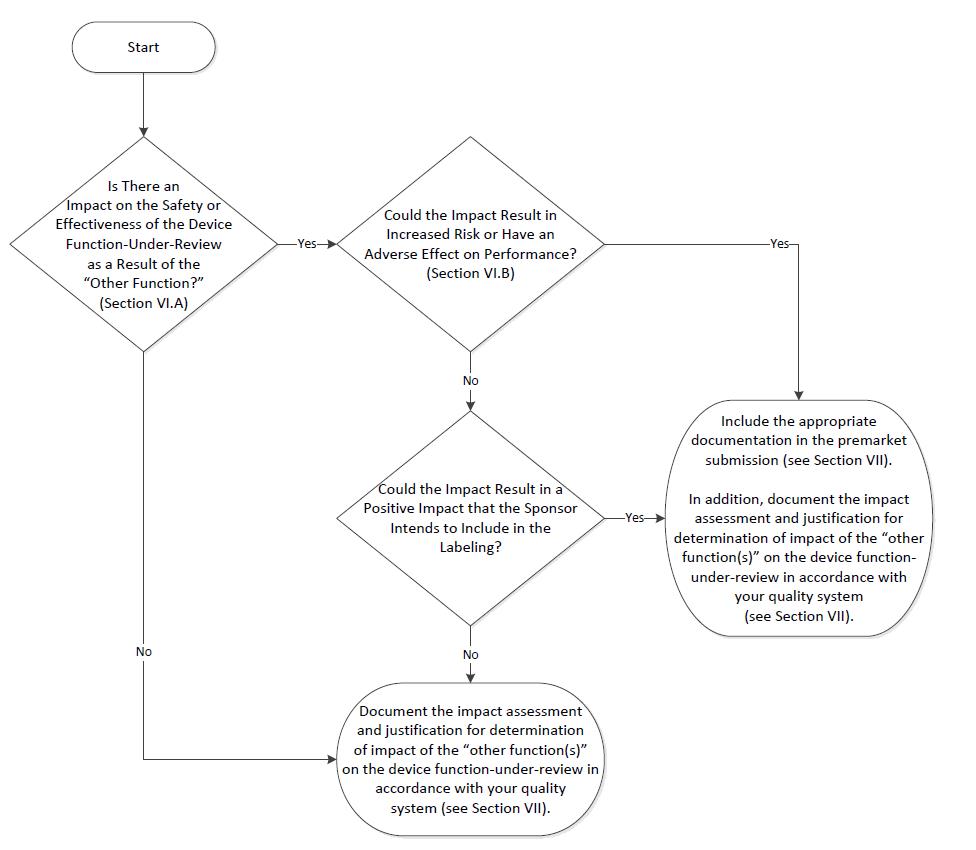
Assessing the Impact of “Other Functions” on the Device Function Under Review 🔗
In the premarket review of a device function-under-review, FDA may assess the impact of “other functions” on the device function-under-review. The premarket assessment of a product with multiple functions is summarized in a two-step process:
(A) Is there an impact on the safety or effectiveness of the device function-under-review as a result of the “other function?;” and, if there is an impact,
(B) Could the impact result in increased risk or have an adverse effect on performance of the device function-under-review, i.e., negative impact?
Quality Management System Considerations 🔗
According to FDA Design Control Requirements, manufacturers must set up procedures for validating their device design, including software validation and risk analysis, where needed. For products with multiple functions, the FDA suggests this validation and risk analysis should include assessing the impact of "other functions" on the device function-under-review. This assessment should take into consideration items A and B from the previous section.
During an inspection, the FDA may request design validation documents, such as impact assessments of "other functions."
Content of a Premarket Submission for Device Function Under Review 🔗
If the "other functions" don’t impact the device function-under-review, then the premarket submission doesn't need to include documentation for the "other functions."
If the "other functions" could negatively impact the device function-under-review, the submission should include the relevant documentation.
If the "other function" has a positive impact on the device function-under-review and the manufacturer wants the FDA to consider it, then the submission should include the necessary documentation. If the positive impact isn't considered a labeled positive impact, the submission doesn't need to include the documentation for the "other function."
The following information should be included in the premarket submission for a MFDP:
Indications for Use 🔗
The indications for use should include only the indications for use of the device function under-review. The indications for use should not include the indications for use of any “other function(s),” unless the sponsor would like the positive impact to be considered in FDA’s assessment of the device function-under-review.
Device Description – Description of Functions 🔗
The device description should explain the device's functions. For products with multiple functions, the description should include details about "other functions" that might negatively impact the device function-under-review, along with how each "other function" affects the main function. If the "other function" has a positive impact on the device function-under-review and is mentioned in the labeling (labeled positive impact), the description should include this information as well.
Sponsors can also describe "other functions" that don't impact or have a positive effect not mentioned in the labeling, to give a better understanding of how the device functions overall.
Labeling 🔗
For products with multiple functions, the labeling should describe the "other functions" sufficiently to ensure the device is used correctly. When creating labeling for a multiple function device product, you may need to include extra information or limitations (such as contraindications, warnings, precautions, adverse reactions) associated with the "other functions" or other instructions specific to your device's design, features, and performance.
Architecture and Design 🔗
When the device function-under-review or "other function" involves software, you should include architecture and design documents suitable for the software's level of concern in the premarket submission. This can be found in the "Guidance for the Content of Premarket Submissions for Software Contained in Medical Devices." The documents should provide enough detail to understand if and how the "other function(s)" interact with or affect the device function-under-review. For instance, an architecture diagram might show the independence of the device function-under-review from the "other function," or design documents might illustrate the use of shared resources.
Device Hazard Analysis 🔗
The device hazard analysis in the premarket submission for the device function-under-review should cover the results of a risk-based evaluation of any potential negative or labeled positive impacts of the "other function" on the safety or effectiveness of the device function-under-review. This assessment should document any risk mitigation strategies used to reduce increased risk or negative effects on performance caused by the combination of the "other function" and the device function-under-review. For instance, if the "other function" might lead to reduced performance of the device function-under-review, documentation should include the risk management process results that identify and describe potential hazards and any necessary risk mitigations employed. You can carry out this risk-based assessment using the process outlined in ANSI/AAMI/ISO 14971: Medical devices – Application of risk management to medical devices, or the Device Hazard Analysis from "Guidance for the Content of Premarket Submissions for Software Contained in Medical Devices."
Requirements and Specifications 🔗
Documentation of requirements and specifications included in the premarket submission for the device function-under-review should include adequate detail to describe any expected relationship, utility, reliance, or interoperability with any “other function.” Software requirements documentation should describe the functional and non-functional requirements of the software system, as identified in the “Guidance for the Content of Premarket Submissions for Software Contained in Medical Devices.”
Performance Testing 🔗
Performance testing for the device function-under-review should take into account any aspects of the "other function(s)" that could impact performance, either negatively or as a labeled positive impact. This will help show that any effects on safety or effectiveness are properly addressed. If you need to conduct safety, performance, or other tests, you should follow the appropriate existing regulations, policies, guidelines, and/or FDA-recognized consensus standards for your device function-under-review.
The full guidance contains additional details and examples related to Multiple Function Device Products.