
The Problem 🔗
When I worked on my first 510(k) submission, I wasted months building documentation that was 10x more complex than necessary. My team and I traced requirements to hazards to risk controls to verification tests in every possible permutation, convinced that more meant better. We created 70-page software development plans filled with process theater. We built elaborate automated test suites for requirements that could've been satisfied with a sentence in the user manual.
I didn't know any better. There were no real examples to learn from—just vague guidance documents and redacted summaries that hid all the useful details. So we over-engineered everything, burned cash, and delayed our timeline by over a year.
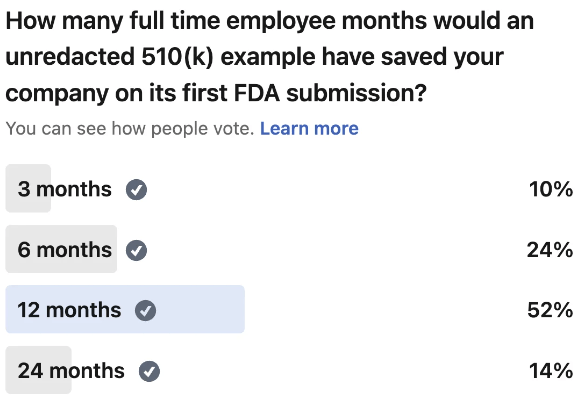
If I'd had access to a some examples back then, I would've saved my team at least 12 months and countless hours of wasted effort. I would've known that a risk management plan could be 7 pages, not 70. That verification protocols didn't need to test everything under the sun. That you could satisfy certain requirements through labeling instead of building complex features.
That's why we're sharing these examples. Because I remember how painful it was to navigate my first submission blind, and I don't want you to go through the same thing.
Here, I’ll walk you through the contents of a 510(k) for an AI/ML enabled software as a medical device (SaMD) for Body Check CT Cardiomegaly (K232613)
Are you allowed to disclose that? 🔗
Our client, Body Check, has granted Innolitics permission to share snippets of the FDA submission to help people just like you. Innolitics takes confidentiality very seriously and will never share such information without written permission.
eSTAR Application 🔗
This document aggregates all the information you will submit to the FDA in a structured way. It also contains “?” that you can click that will provide you some guidance on how to fill out the information. The guidance can be quite vague and having an example will probably save you days to weeks. For instance, do you need to declare conformity to the DICOM standard? We did not. That just saved you thousands of dollars to get a DICOM conformance statement just for the FDA. Your customers may still want it anyway but you can always address the problem when it actually is a problem and you won’t have any customers without FDA clearance first.

Product Development Plan (Software Development Environment Description) 🔗
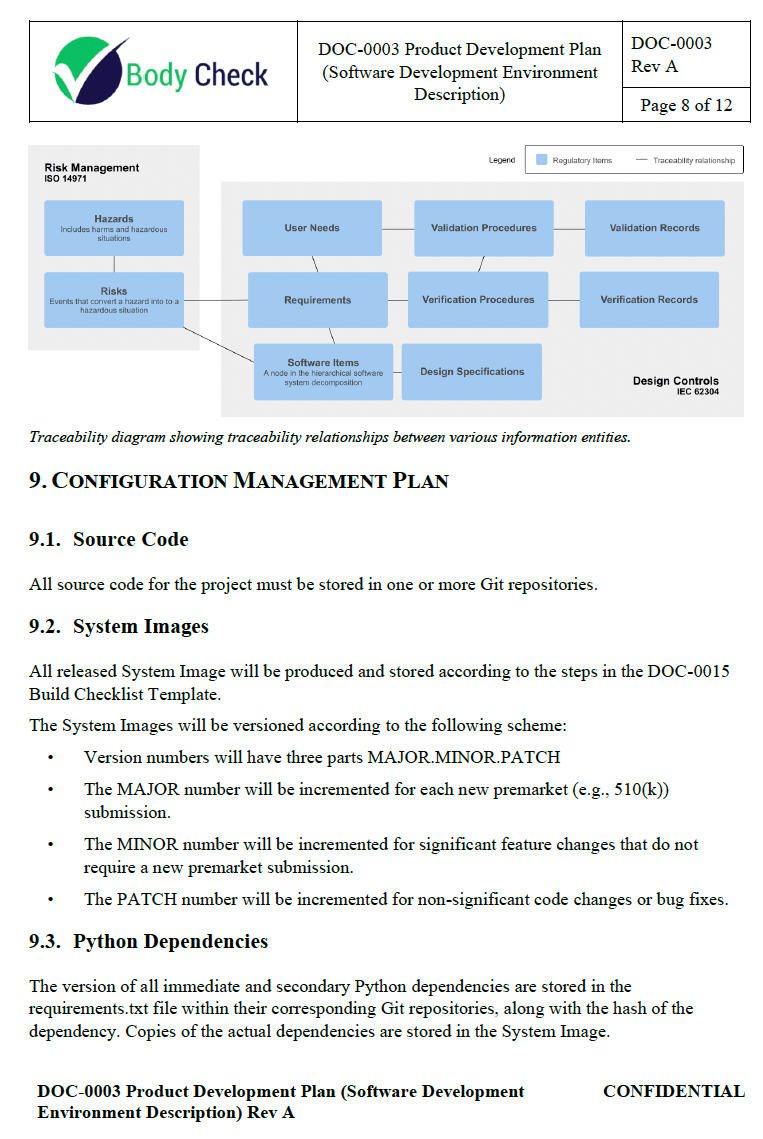
This outlines the plan for developing and bringing the CT Cardiomegaly medical device to market. It covers the scope, definitions, references, procedures, project responsibilities, phases and deliverables, traceability plan, configuration management plan, archival, standards, methods, tools, integration and testing, algorithm development, software verification and validation, and problem resolution.
The first time I inherited such a document from scratch, it was 70 pages long and mostly contained fluff. There is a tendency to think that more is better. All that does is just overwhelm your team with complexity they did not know they have
Risk Management Plan 🔗
This document describes how risk management is planned to be done. Several methods will be used to identify safety risks, including reviewing MAUDE alerts and the FDA Total Product Life Cycle database for related product codes, performing FMEAs, identifying usability risks, and evaluating safety-related device characteristics. Safety risk likelihood is classified into 5 qualitative levels from improbable to frequent. Severity is classified into 5 levels from negligible to catastrophic. Acceptability is determined using a risk matrix based on the likelihood and severity, with risks classified as acceptable, attempt to mitigate, or unacceptable
The first time I prepared a 510(k), seeing a concrete example of this would have probably saved my team 4 months full time engineering time wasted in over-doing it. Heck, even knowing an FDA approved risk management plan was only a total of 7 pages would have probably saved me a month.

System and Software Architecture Diagram 🔗

The purpose of the architectural design chart is to provide a clear roadmap of the device design, showing the software items and layers, their relationships, data inputs/outputs and flow, and how users or external products interact with the system.
Due to confidentiality agreements, I'm unable to share this specific architectural diagram. However, check out our related articles to see how to structure software architecture for medical devices, including best practices for data flow, modular design, and external system interfaces.
Software Requirements Specification 🔗
When I first started working on the software requirements specification for a medical device, I was overwhelmed by the amount of detail and organization required.
This document effectively communicates the key requirements for the CT Cardiomegaly software. It covers functional, performance, interface, security, and regulatory requirements in a concise manner. Due to confidentiality agreements, I'm unable to share this specific architectural diagram. However, check out our related article on the topic:

Risk Assessment 🔗
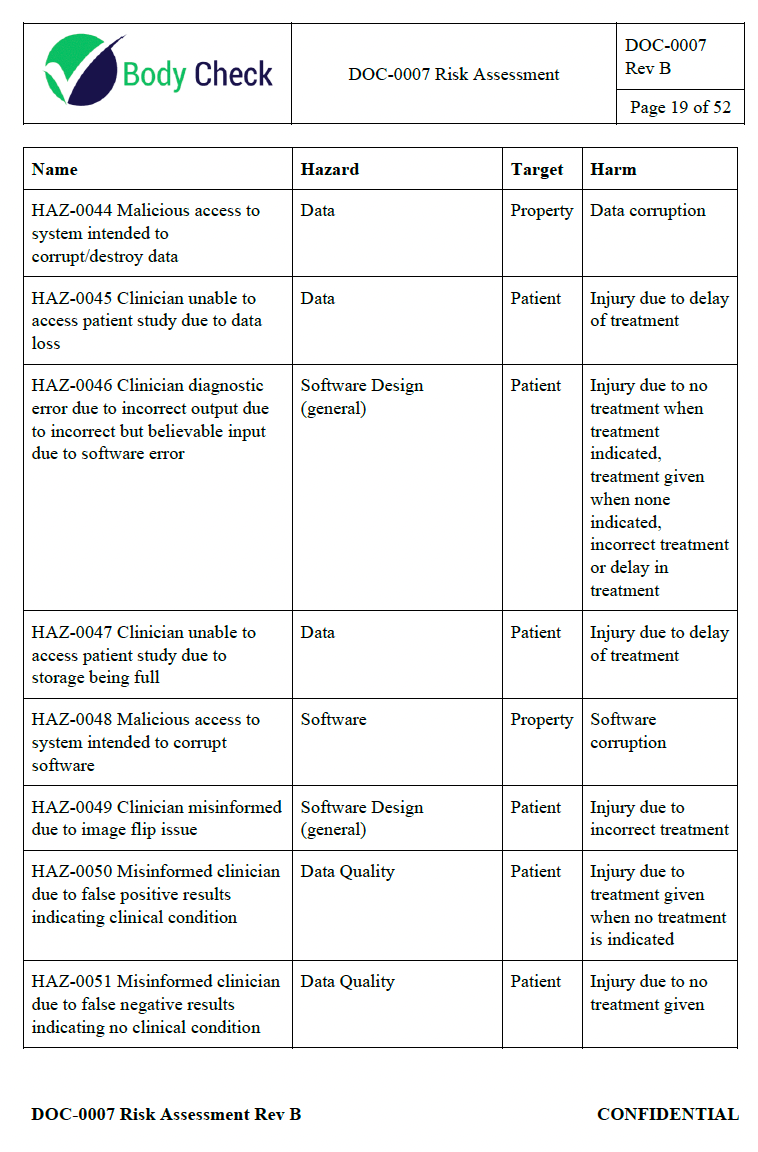
This 52-page document comprehensively covers potential hazardous situations, from compromised data quality due to various biases in the training data, to software design errors, compatibility issues, and even malicious system access. Each risk is systematically analyzed, including the sequence of events leading to the hazardous situation, the severity and likelihood of harm, and the resulting acceptability level.
Here is a related article on the topic:
User Manual 🔗
What goes into a user manual? Good question. There are a lot of examples online but I never understood how it all fit into the overall submission. Did you know that you can have requirements “implemented” in the user manual and verification tests that just check for the presence certain verbiage in the user manual? Having known this alone would have saved my team a cumulative of 8 months over the course of 5 years of writing and maintaining laborious automated tests that could have just as easily been a checkbox.
For instance, we had a requirement stating that the user should be informed about the device's contraindications. We wrote a complex test script to simulate various contraindicated scenarios and ensure that the software behaved accordingly. Had we known that we could satisfy this requirement by simply including a clear list of contraindications in the user manual, we could have saved weeks of development and testing time.

Risk Management Report 🔗

The Risk Management Report is a document that summarizes the risk management activities and conclusions for the CT Cardiomegaly software medical device. Its purpose is to document the device's safety-related characteristics, confirm that the overall residual risk is acceptable, ensure that appropriate methods are in place for collecting and reviewing production and post-production information, and verify that the Risk Management Plan has been properly implemented.
Substantial Equivalence Discussion 🔗

The substantial equivalence discussion focuses on several key areas, such as the devices' intended use, target population, technological characteristics, and performance testing. The document provides a detailed comparison table that outlines the specific characteristics of each device, such as the input, output, measurements, report structure, software requirements, and algorithm type.
Cover Letter 🔗
Did you know that if you filled this form out incorrectly the FDA can release all your documents to the public? If not, you might want to double check.

Software Label 🔗
This is a one pager pointing to the user manual. Some have made the mistake of duplicating the information here which makes it difficult to update the information if it exists in multiple different places.

Device Description 🔗

Overall, the Device Description document provides a clear and detailed overview of the software, its intended use, and its role in assisting healthcare professionals in the detection and assessment of cardiomegaly and related conditions. The contents of this document are typically very similar to the section found in the 510(k) summary and other documents.
Software Description 🔗
The CT Cardiomegaly Software Description document is structured to provide a comprehensive overview of the software's features and functionality. It begins with the Purpose section, outlining the operationally significant features of the software. The Scope section specifies the software version to which the document applies. Following this, the Definitions section lists and explains key terms and abbreviations used throughout the document.
Software Operation describes the software operators, intended patient population, data-analysis methodology, and includes a high-level algorithm data flow diagram. The
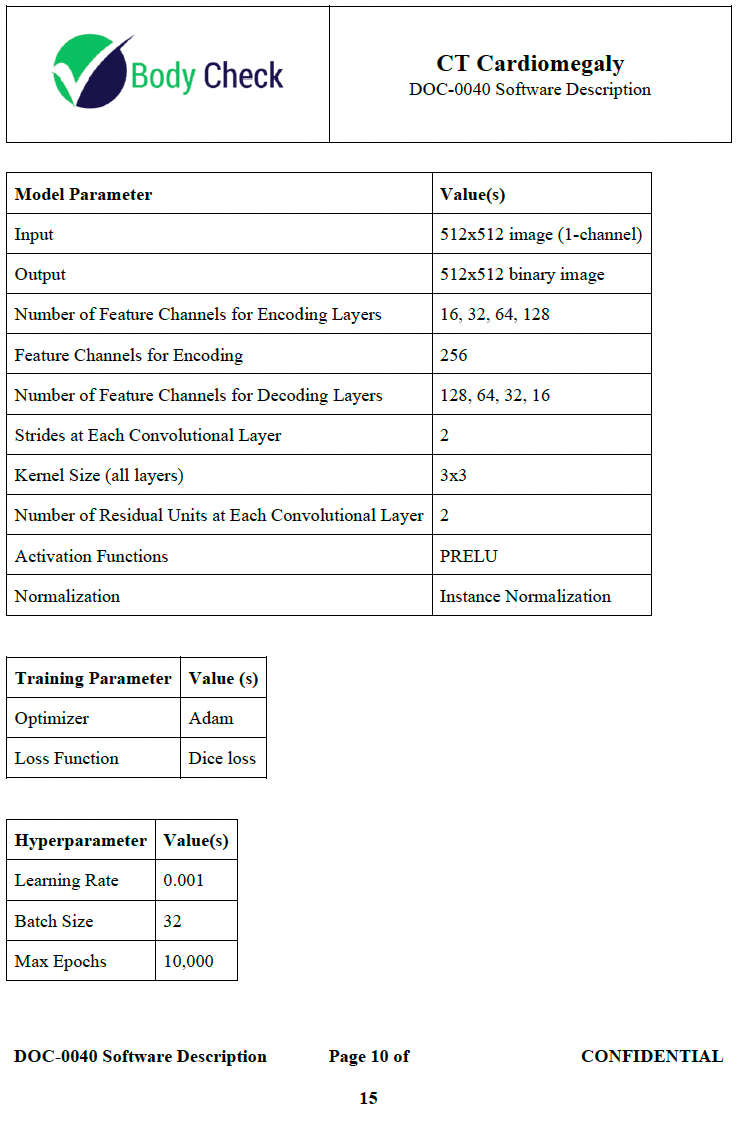
Machine Learning Models section covers the slice localizer, heart segmentation, and inner chest segmentation models, including their architecture, training parameters, and hyperparameters.
Finally, Software Inputs and Outputs describes the inputs and their format, who provides the inputs, device interoperability, outputs and their format, performance testing, intended audience for outputs, data flow, network device interaction, and the use of cloud or network storage.
Verification Plan and Report 🔗

Each verification activity is broken down into detailed subsections:
Procedure: Steps to complete the test, with checkboxes for tracking progress.
Description: A brief overview of what the test entails.
Prerequisites: Requirements or conditions that must be met before the test can be performed.
Test Steps: Specific actions to be taken during the test.
Acceptance Criteria: Conditions that must be satisfied for the test to be considered successful.
Comments: Space for additional observations or notes.
Software Version History 🔗
Some people are afraid of just having one version in their initial FDA application. Well, we proved it can be done. This document was a one pager.
SOUP and OTS Report 🔗
Is your SOUP document shaping up to be hundreds of pages long? You are probably overdoing it. We got ours down to just 7.
Traceability Analysis 🔗
There is very little guidance on what this should actually look like. On my first submission I traced everything to everything, having multiple permutations of hazard to risk to risk control measure to requirement to verification etc only to be frustrated when the matrix did not fit on a landscape 8x11in paper that was needed to get it into a submittable format. The traceability analysis has the potential to be a enormous time sink. Ours’ was only 5 pages long and got cleared with no FDA pushback.
Unresolved Software Anomalies 🔗
There is a common misconception that software needs to be free of defects before FDA submission. It is not true because we had discovered a defect but decided labeling based mitigation was sufficient.
Clinical Performance Assessment Protocol 🔗
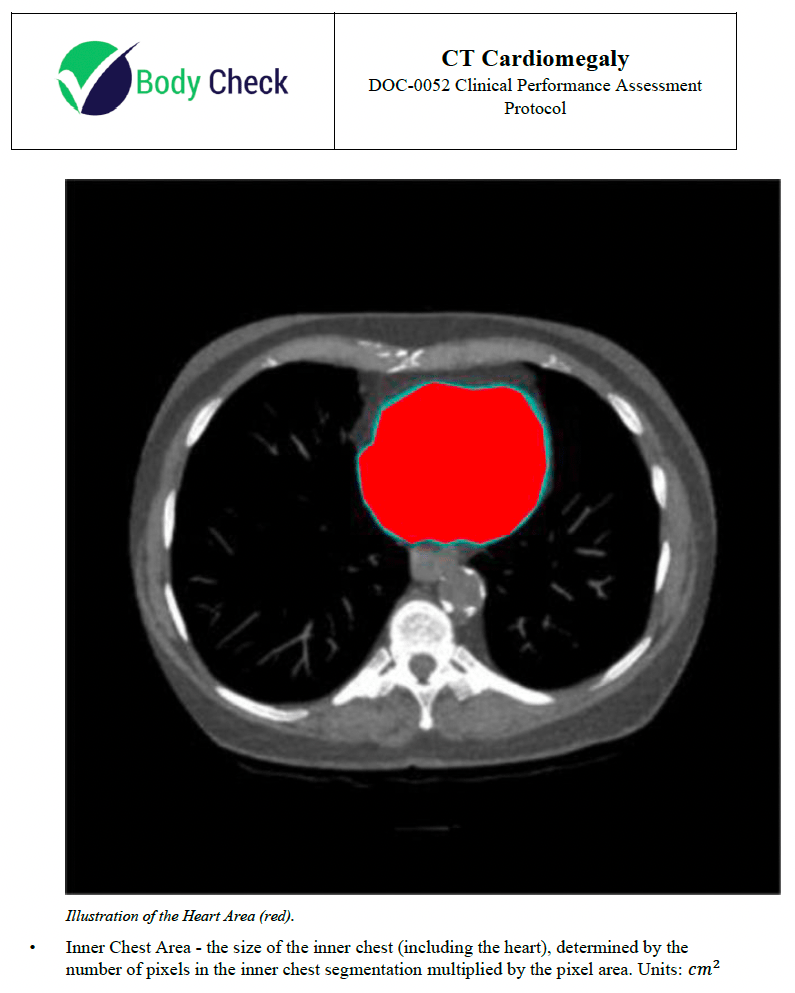
The CT Cardiomegaly Clinical Performance Assessment Protocol document validates the software's intended use by detailing the objectives, scope, definitions, and references. It describes the supported image acquisition devices, algorithm development plan, and data sampling methods. The document outlines inclusion/exclusion criteria, establishes reference standards, and details the measurements for evaluation. It specifies the clinical performance assessment plan, including primary and secondary endpoints, acceptance criteria, and sample size justification. The results section will compare the software's performance against human readers, focusing on key metrics like the Cardiothoracic Ratio and segmentation accuracy.
Clinical Performance Assessment Report 🔗
This document describes the results of the clinical performance assessment protocol. The protocol describes “how” to produce the report. Another common misconception is your device needs to meet all secondary performance metrics. In fact, the device did not meet all of its secondary metrics but passed all of its primary ones. You just need to justify why the device is still safe and effective, which is a better practice than changing our metrics to suit the output of the device.
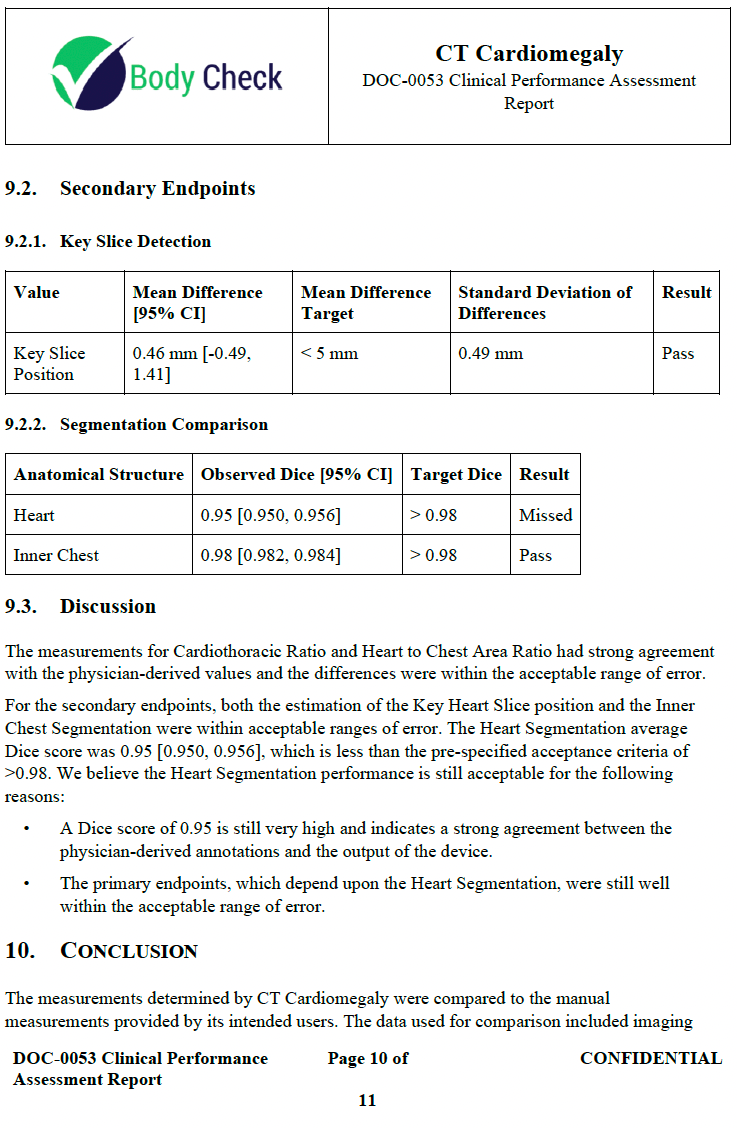
Truthful and Accuracy Statement 🔗
This is just a one pager attesting everything is truthful and accurate. It is extremely important to be completely truthful to the FDA about everything. Don’t overcomplicate this. A one pager is fine.
MDUFMA Cover Sheet 🔗

Every FDA submission must have proof that you paid the FDA’s user fee. This document proves that. In fact, you can pay the MDUFMA fee well in advance of actually submitting the application. The price will only go up because of inflation so you may want to make a payment before the fee goes up on October 1 of almost each year.
Fun fact: During the government shutdown in October of 2025, the government was unable to process payments so this one page is what blocked new FDA submissions that require payments (like a 510k)
Package Labeling Justification 🔗
Does not apply for software as a medical device. Just say “_____ is a Software as a Medical Device, and therefore, has no physical form that requires a package label.”
K232613 Additional Information AINN 🔗

About 3 months after the 510k is submitted, the FDA will send back a letter with a set of findings that you must address in 180 days. In this case, the primary concerns include the need for additional performance testing details, such as Bland-Altman plots and mean absolute error for accuracy assessment, histograms of Dice scores, and the inclusion of Hausdorff distance for segmentation evaluation. There are also requests for clarification on the reference standard, detailed workflow descriptions, and specific testing and validation methods. Additionally, the FDA did not buy our attempt to argue the software was not a “cyber device” so they requested more information on cybersecurity measures, software verification activities, and labeling updates to provide comprehensive user guidance and ensure regulatory compliance.
AINN Clarification Meeting Agenda 🔗
After you receive the AINN, you can request a teleconference with the agency within 10 days. I highly recommend you do this because you can actually resolve quite a few deficiencies that were just due to misunderstandings or oversight. A common mistake is to assume the FDA is omniscient and has seen everything about your application. Assume they are human and humans can make mistakes, skim too fast, go down rabbit holes because of confirmation bias, etc. In this document you can see how we pushed back on FDA’s concerns and you can apply the same principles to your submission too.

Cybersecurity Management Plan 🔗
The CT Cardiomegaly Cybersecurity Management Plan details managing cybersecurity risks throughout the software lifecycle. It covers monitoring and addressing vulnerabilities, creating a software bill of materials (SBOM), and providing updates. Using the STRIDE methodology, the plan includes threat modeling, risk verification, and monthly vulnerability reviews via GitHub’s Dependabot. Critical vulnerabilities are prioritized for resolution. User guidance includes recommended cybersecurity controls and prompt communication of significant risks. Validated software updates follow the P-0002 Development Procedure. The SBOM, in SPDX format, lists all software dependencies, ensuring comprehensive cybersecurity management.

Clinical Performance Assessment Report Rev C 🔗
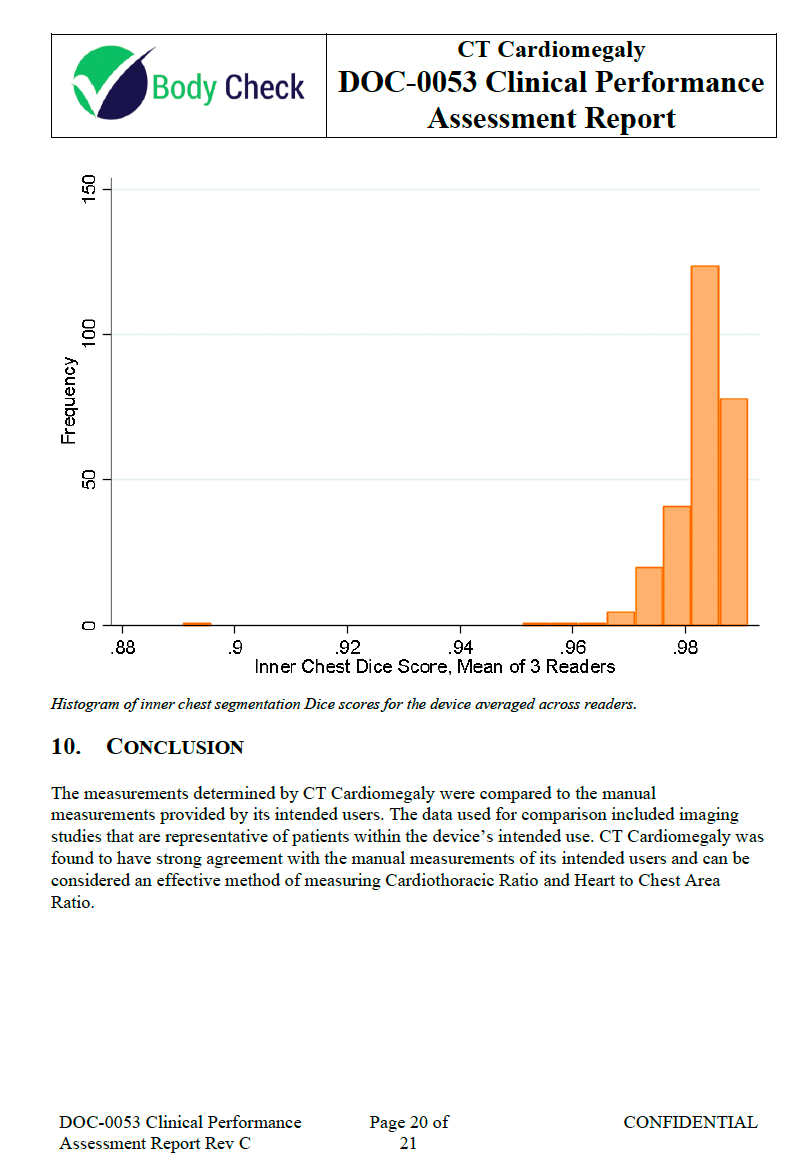
The FDA requested additional analysis to be performed on the data so we performed the analysis and updated the report.
Interactive Review 🔗
Congratulations! This almost certainly means you are close to the finish line. The FDA has determined that your application is close enough to clearance that they don’t need to put you on another hold. They are ok with letting their 90 day clock run instead of pushing the timer on to you. This means they will be quite demanding on your time though. They sent us emails on Friday with a response required by Monday several times during this process.

Need help? 🔗
I hope this article gave you everything you need to get started with your 510(k).
If you would rather just hire us to do it for you, let’s chat today. Our services come with speed and certainty guarantees.




