
Innolitics Introduction 🔗
This FDA guidance is especially important for SaMD products. Proper use of this guidance can reduce the regulatory overhead of your software development.
To best take advantage of this guidance, it helps to have an architectural plan that keeps “other functions” cleanly split from device functions. This is a place where working with a regulatory team that knows software can be beneficial (see Get to Market Faster with Software-Savvy Regulatory Experts).
About this Transcript 🔗
This document is a transcript of an official FDA (or IMDRF) guidance document. We transcribe the official PDFs into HTML so that we can share links to particular sections of the guidance when communicating internally and with our clients. We do our best to be accurate and have a thorough review process, but occasionally mistakes slip through. If you notice a typo, please email a screenshot of it to Miljana Antic at mantic@innolitics.com so we can fix it.
Preamble 🔗
Document issued on July 29, 2020.
For questions about this document regarding CDRH-regulated devices, contact the Division of Digital Health at DigitalHealth@fda.hhs.gov. For questions about this document regarding CBER-regulated devices, contact the Office of Communication, Outreach and Development (OCOD), by calling 1-800-835-4709 or 240-402-8010, or by email at ocod@fda.hhs.gov. For questions about this document regarding CDER-regulated products, contact the Center for Drug Evaluation and Research, Food and Drug Administration, 10903 New Hampshire Ave., Bldg. 51, Rm. 6158, Silver Spring, MD 20993-0002, 301-796-8936. For questions about this document regarding combination products, contact the Office of Combination Products at combination@fda.gov.
Contains non-binding guidance.
I. Introduction 🔗
Medical products may contain several functions, some of which are subject to FDA’s regulatory oversight as medical devices, while others are not. Section 3060(a) of the 21st Century Cures Act (Cures Act) amended the Federal Food Drug, and Cosmetic Act (FD&C Act) to add section 520(o), which excludes certain software functions from the definition of device in section 201(h) of the FD&C Act. The Cures Act also states that in the case of a product with multiple functions that contains both a software non-device function and a device function, FDA may assess the impact that the software non-device function has on the device function when assessing the safety and effectiveness of the device function (section 520(o)(2) of the FD&C Act). FDA believes that a similar approach should be used for the assessment of all multiple function device products.
Products with at least one device function and at least one “other function” (see Section III) are referred to in this guidance as “multiple function device products.” For purposes of this guidance, for any given product, the term “function” is a distinct purpose of the product, which could be the intended use or a subset of the intended use of the product.1 This guidance explains FDA’s regulatory approach and policy for all multiple function device products. Specifically, this guidance clarifies when and how FDA intends to assess the impact of “other functions” that are not the subject of a premarket review on the safety and effectiveness of a device function that is subject to FDA review. The purpose of this guidance is to identify the principles, premarket review practices, and policies for FDA’s regulatory assessment of such products, and to provide examples of the application of these policies.
For the current edition(s) of the FDA-recognized consensus standard(s) referenced in this document, see the FDA Recognized Consensus Standards Database.2 For more information regarding use of consensus standards in regulatory submissions, please refer to the FDA guidance titled “Appropriate Use of Voluntary Consensus Standards in Premarket Submissions for Medical Devices”3 and “Standards Development and the Use of Standards in Regulatory Submissions Reviewed in the Center for Biologics Evaluation and Research.”4
FDA’s guidance documents, including this guidance, do not establish legally enforceable responsibilities. Instead, guidances describe the Agency’s current thinking on a topic and should be viewed only as recommendations, unless specific regulatory or statutory requirements are cited. The use of the word should in Agency guidance means that something is suggested or recommended, but not required.
II. Background 🔗
Section 201(h) of the Federal Food, Drug, and Cosmetic Act (FD&C Act) defines a device as:
an instrument, apparatus, implement, machine, contrivance, implant, in vitro reagent, or other similar or related article, including any component, part, or accessory, which is:
- recognized in the official National Formulary, or the United States Pharmacopoeia, or any supplement to them,
- intended for use in the diagnosis of disease or other conditions, or in the cure, mitigation, treatment, or prevention of disease, in man or other animals, or
- intended to affect the structure or any function of the body of man or other animals, and
which does not achieve its primary intended purposes through chemical action within or on the body of man or other animals and which is not dependent upon being metabolized for the achievement of its primary intended purposes. The term "device" does not include software functions excluded pursuant to section 520(o).5
Functions that fall within this definition are devices and, therefore, subject to FDA’s oversight. FDA’s regulation of devices is tailored based on risk. For example:
- Some devices are subject to premarket review, either through a premarket notification under section 510(k) of the FD&C Act, a premarket approval (PMA) application under section 515 of the FD&C Act, a De Novo classification request under section 513(f)(2) of the FD&C Act, an investigational device exemption (IDE) application under 21 CFR 812, or a humanitarian device exemption (HDE) under section 520(m) of the FD&C Act. Certain devices are subject to premarket review through a biological license application (BLA) under section 351 of the Public Health Service Act.
- Other, lower-risk devices are exempt from premarket review, but are subject to general controls, including registration and listing,6 good manufacturing practices,7 and adverse event reporting.8
- FDA has issued guidance that it does not intend to focus its regulatory oversight on some devices that pose a low risk to patients. FDA has done this for many low-risk software functions. See, for example, FDA’s guidance “Policy for Device Software Functions and Mobile Medical Applications”9 and “General Wellness: Policy for Low Risk Devices.”10
On December 13, 2016, the Cures Act was signed into law. Section 3060(a) of this legislation, titled “Clarifying Medical Software Regulation,” amended the FD&C Act to add section 520(o), which describes software functions that are excluded from the definition of the term device in section 201(h). In addition, section 520(o)(2), reproduced below, describes the regulation and assessment of a software product with multiple functions, including at least one device function and at least one software function that is not a device.
Section 520(o)(2) of the FD&C Act (21 U.S.C. 360j(o)(2))
In the case of a product with multiple functions that contains—
(A) at least one software function that meets the criteria under paragraph (1) or that otherwise does not meet the definition of device under section 201(h); and
(B) at least one function that does not meet the criteria under paragraph (1) and that otherwise meets the definition of a device under section 201(h),
the Secretary shall not regulate the software function of such product described in subparagraph (A) as a device. Notwithstanding the preceding sentence, when assessing the safety and effectiveness of the device function or functions of such product described in subparagraph (B), the Secretary may assess the impact that the software function or functions described in subparagraph (A) have on such device function or functions.
III. Scope 🔗
As mentioned above, in this guidance for any given product, the term “function” is a distinct purpose of the product, which could be the intended use or a subset of the intended use of the product. For example, a product with an intended use to analyze data has one function: analysis. A product with an intended use to store, transfer, and analyze data has three functions: (1) storage, (2) transfer, and (3) analysis. As this example illustrates, a product may contain multiple functions.
The following terms are used for the purposes of this guidance:
- Device function: function that meets the definition of a device under section 201(h) of the FD&C Act.
- “Other function:” function that:
- does not meet the definition of device;
- meets the definition of device, but is not subject to premarket review (e.g., 510(k)-exempt11); or
- meets the definition of device, but for which FDA has expressed its intention not to enforce compliance with applicable regulatory controls.
- Device function-under-review: function for which FDA is conducting premarket review.
A multiple function device product contains at least one device function and at least one “other function.” In the premarket review context (see Sections IV-VIII), a multiple function device product is a product with at least one device function-under-review and one “other function.” For example, during premarket review of a multiple function device product, a device function that requires a PMA would be evaluated according to the principles in this guidance. In the postmarket context (see Section IX), a multiple function device product is a product with at least one device function that is the focus of FDA’s oversight and one “other function.” For example, during an inspection of a facility manufacturing a multiple function device product, the principles of this guidance would apply to a 510(k)-exempt device function. In accordance with existing policies, FDA intends not to review a device function subject to an enforcement discretion policy merely because it is part of a multiple function device product.12
Although section 520(o)(2) of the FD&C Act applies to the regulation of products containing at least one device function and at least one non-device software function, FDA believes the same principles should apply to the assessment of all multiple function device products, whether those functions are software-based, hardware-based, or both. This document does not provide guidance on which functions do and do not meet the definition of a device. It also does not provide guidance on which functions meet the device definition but are those for which FDA has expressed its intention not to enforce compliance with applicable requirements of the FD&C Act.
Sections IV-VI and VIII-IX of this document discuss the policy in this guidance for multiple function device products, as well as modifications to “other functions” and postmarket requirements. Generally, these policies (in Sections IV-VI and VIII-IX) apply to device functions-under-review within a multiple function device product, including multiple function device products included in a combination product.13 Section VII addresses the premarket submission content for device functions-under-review of a multiple function device product; this approach also applies to multiple function device products that are part of a device-led combination product. Similar principles may apply for drug- or biologic-led combination products that include a multiple function device product. This guidance does not address the premarket submission requirements, nor submission requirements for modifications, of a drug or biologic constituent part of a combination product. This guidance does not change FDA’s review of a drug or biologic constituent part of a combination product.
The principles outlined in this guidance reflect a safety-based approach to risk management aligned with ANSI/AAMI/ISO 14971: Medical devices – Application of risk management to medical devices.
IV. Policy: Premarket Review of Multiple Function Device Products 🔗
Consistent with the FD&C Act, as amended by the Cures Act, certain software functions in a multiple function device product are not a device because they do not meet the statutory device definition in section 201(h) of the FD&C Act.14 Such “other functions” are not the subject of FDA’s review simply because they are part of a multiple function device product. However, FDA may assess the impact of “other functions” when assessing the safety and effectiveness of the device functions-under-review of a multiple function device product.15
For example, FDA does not regulate a general-purpose computing platform, but may assess its impact on the safety and effectiveness of a device function-under-review, such as a mobile medical application.16 This approach is similar to how FDA considers a product that includes an intragastric balloon subject to premarket approval and an endoscope accessory that is 510(k)-exempt (e.g., an endoscope guidewire); in this case, FDA may assess the impact of the endoscope accessory on the safety and effectiveness of the intragastric balloon.
For some device functions, FDA has expressed its intention not to enforce compliance with applicable requirements. In accordance with existing policies, FDA intends not to review a device function subject to an enforcement discretion policy merely because it is part of a multiple function device product.17 Instead, FDA intends to review the device function(s) for which clearance or approval is being sought (e.g., the device function-under-review). For example, if a manufacturer seeks clearance or approval for a device function (e.g., analysis), and not the device function for which FDA has expressed its intention not to enforce compliance (e.g., trend), then FDA intends to only review the analysis function and assess the trend function only insofar as it could negatively impact the analysis function. In that instance, because FDA is reviewing the analysis function only, FDA’s decision to clear or approve applies only to the analysis function.
As described in Section VI.A, in their risk assessment process, manufacturers should determine if an “other function” impacts the safety or effectiveness of the device function-under-review. Such impacts can be negative, e.g., “other functions” that may adversely affect the performance of the device function-under-review by slowing computation time, or positive, e.g., “other functions” that enhance safety and effectiveness of the device function-under-review because they improve processing speed. As described in Section VII, for multiple function device products, manufacturers should provide in their premarket submission information that is related to the impacts of “other functions” only if they could negatively impact the device function under-review or if they positively impact the device function-under-review and this fact will be represented in the device function-under-review’s labeling (“labeled positive impact”). For those premarket submissions during which FDA reviews draft labeling, a labeled positive impact should be included in the indications on the CDRH Premarket Review Submission Cover Sheet.18 Although FDA does not recommend submitting information about all positive impacts from “other functions” in a premarket submission, all impact assessments (negative, positive, and no impact) must be performed and documented as part of design validation under 21 CFR 820.30(g) for multiple function device products (see also Section VII) and are subject to FDA review during an inspection.19
V. Considerations for Multiple Function Device Products 🔗
FDA guidances with premarket submission recommendations for medical devices apply to products with at least one device function-under-review. The following sections describe additional considerations for multiple function device products. However, there is no one-size fits-all approach for the wide variety of multiple function device products, and manufacturers should consider their products’ specific functions and conditions of use.
A. Separation in Design and Implementation of the Device Function 🔗
The device function-under-review should, to the extent possible, be separated from “other functions” in its design and implementation (e.g., logical separation, architectural separation, code, and data partitioning). The use of partitions in software systems can be an effective or sometimes necessary strategy to limit hazardous situations and to reduce risk in order to prevent the “other function” from negatively impacting the device function-under-review.
Documenting the results of a thorough risk analysis of the impacts of “other functions” and mitigation strategies employed is a critical component of a risk management process. The higher the degree of separation, the easier it is to independently review the safety and effectiveness of the device function-under-review. Separation will also increase the likelihood that the device function-under-review is not dependent on the “other function(s)” in the product.
Architecture decisions early in the design cycle can facilitate optimal separation and support segregation necessary for risk control. When separation is not achievable, the interconnection and interdependencies between the device function-under-review and “other functions” should be explained and included in the hazard analysis, and appropriate controls should be created to reduce the adverse impact of the connectivity on the safety and effectiveness of the device function-under-review.
When considering cybersecurity risks, some level of separation of the device function-under review from the “other function(s)” in design and implementation may be necessary to mitigate cybersecurity risks to the device function-under-review. For example, modular and/or separation architectures can help reduce the possibility that a cybersecurity threat to/from an “other function” could impact the device function-under-review.
See Sections VI and VII for discussion regarding how to assess the impact of “other functions” on the device function-under-review and what content resulting from this assessment should be included in a premarket submission for the device function-under-review.
B. Impact of “Other Functions” 🔗
The manufacturer of a multiple function device product should consider the following regarding all “other functions” of the product:
- the role of the “other function” in the device function-under-review’s performance;
- any limitations of the device function-under-review when using the “other function;”
- developing appropriate hardware and software resource specification(s) for the product with multiple functions to ensure minimal impact of the “other function” on the performance of the device function-under-review;
- how to ensure appropriate actions are taken by the end user when using the device function-under-review with the “other function;” and
- identification, evaluation, and mitigation of any additional risks, including cybersecurity risks, in the device function-under-review when used in combination with the “other function.”
VI. Assessing the Impact of “Other Functions” on the Device Function Under Review 🔗
In the premarket review of a device function-under-review, FDA may assess the impact of “other functions” on the device function-under-review. The premarket assessment of a product with multiple functions is summarized in a two-step process:
(A) Is there an impact on the safety or effectiveness of the device function-under-review as a result of the “other function?;” and, if there is an impact,
(B) Could the impact result in increased risk or have an adverse effect on performance of the device function-under-review, i.e., negative impact?
The flowchart below is intended as a guide to be used with the textual descriptions in this guidance.
Each decision point in the flowchart and its relevant considerations are described in the Sections below. Section VII describes what should be included in the documentation for a premarket submission for a multiple function device product if the device function-under-review is impacted by the “other function,” and if the impact could result in increased risk or an adverse effect on performance.
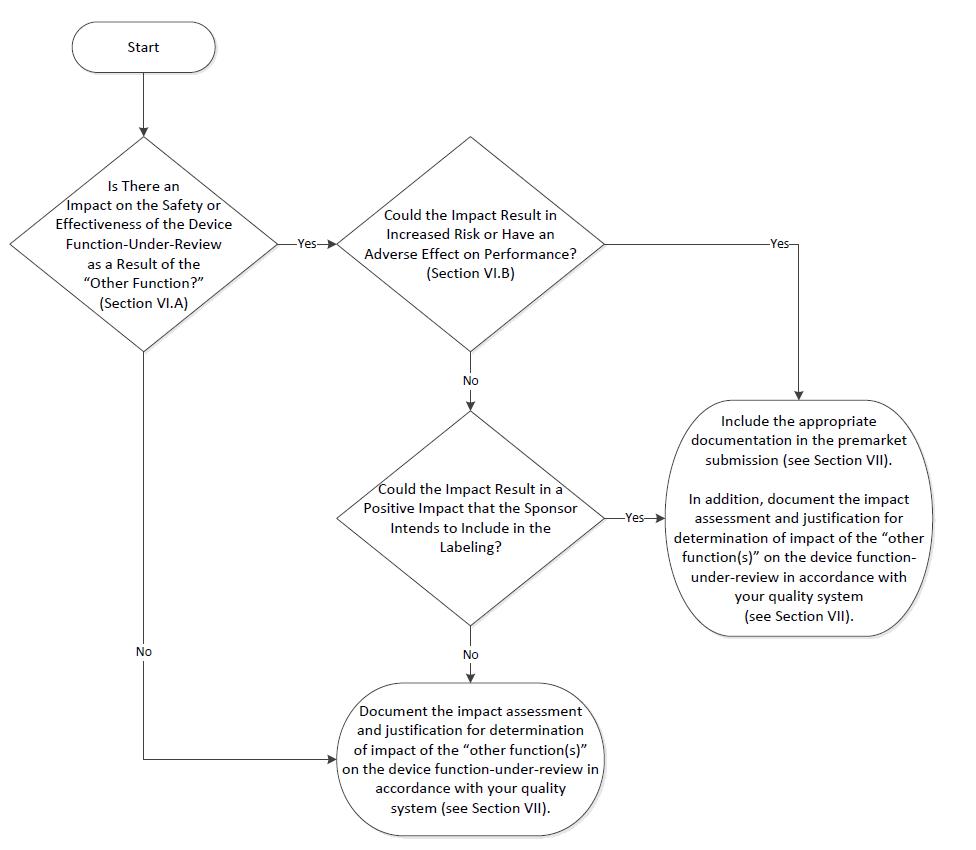
A. Is There an Impact on the Safety or Effectiveness of the Device Function-Under-Review as a Result of the “Other Function?” 🔗
Manufacturers should determine if an “other function” impacts the safety or effectiveness of the device function-under-review. If so, FDA recommends that manufacturers evaluate that impact and include the relevant parts of the “other function” in their hazard analysis.
When assessing the impact of “other functions” on the device function-under-review, it is important to consider the various relationships between the functions that may exist in a multiple function device product. The existence of a relationship does not necessarily mean that there may be an impact on the safety or effectiveness of the device function-under-review. For example, a software device function, including a mobile medical application, has a relationship with the computing platform on which it is executed. The computing platform may or may not have an impact on the safety or effectiveness of the software device function. See the examples in Appendix 1: Examples of Multiple Function Device Products.
When assessing if an “other function” impacts the device function-under-review within the same product, considerations should include whether there are shared computational resources, data dependencies, or any other type of relationship between the functions. The following are examples of questions that may help to determine if an “other function” may impact the safety or effectiveness of the device function-under-review. Note that this is not intended to be a comprehensive list of considerations, and manufacturers should conduct their own risk assessments.
- Does the “other function” provide input data that is used for a critical calculation within the device function-under-review?
- Does the device function-under-review rely on results from the “other function?”
- Do the “other function” and the device function-under-review share code that is necessary for proper execution of the device function-under-review?
- Does the “other function” write to memory or other storage that stores code or data of the device function-under-review? (E.g., does the “other function” directly modify the configuration of the device function-under-review, bypassing the designed input and output methods?)
- Do the “other function” and the device function-under-review share the same output screen or graphical user interface?
- Does the “other function” affect the processing time necessary for the performance of the device function-under-review when the functions share a processor?
- Does the “other function” affect the memory requirements for the performance of the device function-under-review when the functions share memory on the computer platform?
- Do the “other function” and the device function-under-review share programming pointers?
- Does the device function-under-review need privileges to prevent delays or interruptions that may result from the “other function?”
B. Could the Impact Result in Increased Risk or Have an Adverse Effect on Performance, i.e., a Negative Impact? 🔗
If the “other function” impacts the device function-under-review, the extent of the impact should be evaluated and included in the hazard analysis. The manufacturer should determine whether the impacts could be positive or negative. For positive impacts, the manufacturer should identify the beneficial impact that the “other function” has on the device function-under-review when operating as intended and confirm that there is no adverse impact on the device function-under-review if the “other function” were to fail to operate as intended. For negative impacts, the manufacturer should identify whether there could be increased risk and/or an adverse effect on performance due to the combination of the “other function” with the device function-under-review.
1. Impacts to Safety. 🔗
The impacts to safety are often impacts associated with risk. A risk-based assessment should be used to identify and analyze all risks of a device function-under-review, including those that may result from the inclusion of “other functions” in the product. If the impact results in no increased risk, then no additional risk mitigation is necessary. If there could be increased risk, then the risk should be appropriately documented and mitigated, and the appropriate verification and/or validation should be performed to ensure the effectiveness of the mitigation. The following examples can be used as a guide to understand increased risk.
- The “other function” introduces a new hazardous situation or a new cause of an existing hazardous situation that is not otherwise present in the device function-under-review.
- A “hazardous situation” exists when there is exposure to a hazard (i.e., a potential source of harm) that can lead to physical injury or damage to the health of people.
- The “other function” increases the severity of harm associated with a hazardous situation identified for a device function-under-review. · The “other function” is a risk control measure for a device function-under-review.
- The use of or implementation of the “other function” impacts a risk control measure for a device function-under-review.
When assessing the cybersecurity risks of “other functions” on the device function-under-review, it should be assumed that the “other function(s)” may be employed (maliciously or unintentionally) to cause an adverse impact on the device function-under-review (e.g., as a result of a cyberattack). For example, the unavailability of the device function-under-review could present risks that should be considered and appropriately mitigated, because a cybersecurity threat to the “other function(s)” may lead to the device function-under-review becoming unavailable to the intended user. Software transparency methods aid in the identification of software dependencies between a device function-under-review and “other function(s),” as well as identify vulnerable software components in “other functions” that can be used to impact device functions.20
2. Impacts to Effectiveness 🔗
The impacts to effectiveness are typically impacts to the performance of the device, for example, impacts to the responsiveness, usability, or efficiency of the device. If there could be adverse impacts to the device function-under-review as a result of the “other function(s),” then appropriate verification and validation should be performed with the product to characterize the performance of the device function-under-review and evaluate if there may be an adverse effect on the performance. The following examples can be used as a guide to determine whether there could be an adverse impact on performance.
- The performance or clinical functionality of the device function-under-review depends on the “other function” for the device function-under-review to perform as specified.
- The performance of the device function-under-review fails to meet the specified performance level due to the “other function.”
Note that the relationship between a device function-under-review and the “other function(s)” may be limited to sharing resources on a general-purpose computing platform. If the only identified relationship between the device function-under-review and an “other function” is the sharing of a common computing platform, the risk assessment for the device function-under-review should include the hazards associated with running on a common general-purpose computing platform. For examples of multiple function device products and explanations of the assessment of how “other functions” impact the device function(s)-under-review, see Appendix 1: Examples of Multiple Function Device Products.
VII. Content of a Premarket Submission for Device Function-Under-Review 🔗
Under 21 CFR 820.30(g), a manufacturer must establish and maintain procedures for validating its device design. Such design validation shall include software validation and risk analysis, where appropriate. For a multiple function device product, FDA recommends that this validation and risk analysis include an impact assessment and rationale for a manufacturer’s determination of the impact of the “other function(s),” whether no impact, negative, or positive, on the device function-under-review in accordance with the manufacturer’s quality system, where appropriate. The impact assessment should include:
- whether or not there is an impact on the safety or effectiveness of the device function-under-review as a result of the “other function,” and if there is,
- whether the impact could result in increased risk or either an adverse or positive effect on performance.
FDA may request to see design validation documents, including, for example, the documentation of the impact assessment of the “other function(s)” on the device function-under-review, during an inspection.21
If the “other function” does not impact the device function-under-review (i.e., there is no impact on the safety and effectiveness of the device function-under-review), then the premarket submission for the device function-under-review does not need to include the documentation described in this section for the “other function,” consistent with FDA’s guidance “The Least Burdensome Provisions: Concept and Principles.”22
If the “other function” could adversely impact the device function-under-review (i.e., the impact of the “other function” may be increased risk or adverse effect on performance of the device function-under-review), then the premarket submission for the device function-under-review should include the appropriate documentation identified below for the “other function.”
If the device function-under-review could be positively impacted by the “other function” (i.e., the impact of the “other function” has a beneficial impact on the device function-under-review when operating as intended and does not have an adverse impact on the device function-under-review if the “other function” fails to operate as intended; for example, the “other function” improves the computational speed of the device function-under-review, and therefore positively impacts the device function-under-review’s performance), and the sponsor would like the positive impact to be considered in FDA’s assessment of the device function-under-review, then the premarket submission for the device function-under-review should include the appropriate documentation identified below for the “other function.” If the device function-under-review could be positively impacted, but is not considered a labeled positive impact, then the submission does not need to include the documentation described in this section for the “other function.”
A. Indications for Use 🔗
The indications for use should include only the indications for use23 of the device function-under-review. The indications for use should not include the indications for use of any “other function,” unless the sponsor would like the positive impact to be considered in FDA’s assessment of the device function-under-review.
B. Device Description – Description of Functions 🔗
The device description should include an explanation of the device’s functions.
For a multiple function device product, the device description should include a description of the “other function(s)” that could adversely impact the device function-under-review and should address how the device function-under-review is impacted by each of the “other functions.”
If the device function-under-review could be positively impacted by the “other function,” and the labeling reflects the positive impact (labeled positive impact), the device description should include the information outlined above in regard to the positive impact of the “other function” on the device function-under-review.
Sponsors may also describe “other functions” that either do not have an impact or could have a positive impact that is not suggested in the labeling of the device function-under-review, to provide an explanation of how the device functions overall.
C. Labeling 🔗
FDA regulates device labeling in several ways. For example, section 502(f) of the FD&C Act requires that labeling include adequate directions for use. Under section 502(a)(1) of the FD&C Act, a medical device is deemed misbranded if its labeling is false or misleading in any particular. Under section 201(n), labeling may be misleading if it fails to reveal facts material with respect to consequences that may result from use of the article under the conditions of use prescribed in the labeling or under such conditions of use as are customary or usual. See also 21 CFR 1.21.
For multiple function device products, the labeling should include a description of the “other function(s)” adequate to ensure appropriate use of the device.
Device labeling must comply with applicable statutes and regulations.24 When developing the labeling for a multiple function device product, it may be necessary to include additional information or limitations (e.g., contraindications, warnings, precautions, adverse reactions) associated with the “other function(s),” or other instructions that are appropriate for your device, depending on its specific design, features, and performance characteristics.
D. Architecture and Design 🔗
When the device function-under-review or “other function” includes software, the architecture and design documents appropriate to the software level of concern should be included in the premarket submission (“Guidance for the Content of Premarket Submissions for Software Contained in Medical Devices”25) for the device function-under-review, and should include adequate detail to understand how or if the “other function(s)” interact with or impact the device function-under-review. For example, an architecture diagram may demonstrate the independence of the device function-under-review from the “other function,” or design documents may demonstrate the use of shared resources.
E. Device Hazard Analysis 🔗
The device hazard analysis included in the premarket submission for the device function-under-review should include the results of a risk-based assessment of any potential adverse impact or labeled positive impact of the “other function” to the safety or effectiveness of the device function-under-review as discussed above. The results of the risk-based assessment should document any risk mitigations employed to mitigate increased risk or adverse effect on performance due to the combination of the “other function” with the device function-under-review. For example, if the impact of the “other function” could result in decreased performance of the device function-under-review, documentation should include the results of the risk management process that identifies and describes the hazards that could affect the safety or could cause decreased performance and any necessary risk mitigations employed. This risk-based assessment could be performed using the process outlined in ANSI/AAMI/ISO 14971: Medical devices – Application of risk management to medical devices, or the Device Hazard Analysis from “Guidance for the Content of Premarket Submissions for Software Contained in Medical Devices,”26 for example.
F. Requirements and Specifications 🔗
Documentation of requirements and specifications included in the premarket submission for the device function-under-review should include adequate detail to describe any expected relationship, utility, reliance, or interoperability with any “other function.” Software requirements documentation should describe the functional and non-functional requirements of the software system, as identified in the “Guidance for the Content of Premarket Submissions for Software Contained in Medical Devices,”27 as appropriate.
G. Performance Testing 🔗
Performance testing for the device function-under-review should be conducted considering those aspects of the “other function(s)” that have an impact (either if there could be an adverse impact or labeled positive impact) on the performance in order to demonstrate that the impact to safety or effectiveness is appropriately addressed. If you need to perform safety, performance, or other testing, you should follow the relevant existing regulations, policies, guidances, and/or FDA recognized consensus standards for your device function-under-review.
H. Submission Summary 🔗
Where the device function-under-review could not be adversely impacted by an “other function,” FDA does not intend to assess that “other function” (unless the sponsor would like FDA to consider the positive impact of the “other function” in FDA’s assessment of the device function-under-review). Therefore, an approved or cleared device may include functionality that FDA has not assessed. FDA intends to make the extent of the product’s assessment clear, such as with a statement in the 510(k) Summary, PMA Summary of Safety and Effectiveness Data (SSED), De Novo Decision Summary, or HDE Summary of Safety and Probable Benefit (SSPB). For example, if a product includes a device function-under-review and an “other function” that is not subject to premarket review, the statement might read:
This medical device product has functions subject to FDA premarket review as well as functions that are not subject to FDA premarket review. For this application, if the product has functions that are not subject to FDA premarket review, FDA assessed those functions only to the extent that they either could adversely impact the safety and effectiveness of the functions subject to FDA premarket review or they are included as a labeled positive impact that was considered in the assessment of the functions subject to FDA premarket review.
VIII. Modifications to the “Other Function” of a Multiple Function Device Product 🔗
If there is a modification to the “other function” of a multiple function device product, then, in accordance with the policy in Section VI, the modification should be assessed to determine if the change could significantly impact the safety or effectiveness of the device function that was the subject of FDA review.28 If you find such an impact on the device function that was the subject of FDA review, and it could adversely impact the device function or positively impact the device function and that impact is included in the device function’s labeling (labeled positive impact), then reference the “Deciding When to Submit a 510(k) for a Change to an Existing Device” Guidance,29 “Deciding When to Submit a 510(k) for a Software Change to an Existing Device” Guidance,30 or “Modifications to Devices Subject to Premarket Approval (PMA) - The PMA Supplement Decision-Making Process” Guidance,31 to help determine if a new premarket submission is necessary for the modification to the “other function” as required by 21 CFR 807.81(a)(3). Unless the positive impact is included in the device function’s labeling (labeled positive impact), if such a modification could positively impact the device function, then FDA does not intend at this time to enforce the applicable premarket submission requirements.
The sponsor should document the impact assessment and justification for their determination of impact of the modification of the “other function” on the device function-under-review in accordance with the sponsor’s quality system. The impact assessment should include whether or not there is an impact on the safety or effectiveness of the device function-under-review as a result of the “other function,” and, if so, whether the impact could result in increased risk or either an adverse or positive effect on performance. If the modification to the “other function” of a multiple function device product is part of a combination product that was submitted under a drug or biologic product application type, the information related to the change should be submitted using the appropriate drug or biologic submission, if required.32
IX. Application of Other Device and Postmarket Requirements 🔗
General control requirements33 apply to device functions subject to 510(k), PMA, De Novo, or HDE requirements and to device functions that are 510(k)-exempt. For example, device functions in multiple function device products must comply with design control requirements under the Quality System regulation (21 CFR Part 820). As another example, in accordance with 21 CFR 803.50, FDA expects the manufacturer of a device function to investigate the cause of an adverse event and to submit an adverse event report when the manufacturer becomes aware of information that reasonably suggests that the device function may have caused or contributed to a death or serious injury, among other circumstances.
If the manufacturer of a multiple function device product determines that an “other function” affected the safety or effectiveness of the device function leading to a reportable death, serious injury, or malfunction under 21 CFR Part 803, or is not certain about whether the device function or an “other function” in a product caused or contributed to a reportable event, the manufacturer would still be required to report it if the information reasonably suggests that the device may have caused the death or serious injury.
FDA continues to intend not to enforce general control requirements for device functions for which FDA has expressed its intention to not enforce applicable regulatory controls at this time.34
Combination products that include multiple function device products generally have additional requirements, such as those requirements applicable to the drug or biologic constituent part. For more information on current good manufacturing and postmarketing safety reporting requirements and policies applicable to combination products, see 21 CFR Part 4, FDA “Current Good Manufacturing Practice Requirements for Combination Products”35 and the Office of Combination Products’ (OCP’s) Postmarketing Safety Reporting for Combination Products webpage.36 Additional information on regulatory requirements and policies for combination products can also be found on OCP’s webpage.37
Appendix 1: Examples of Multiple Function Device Products 🔗
The following are hypothetical examples of multiple function device products, explaining the impact assessment of the “other function” on the device function(s)-under-review. These generalized examples are not intended to cover all possible details, risks, or considerations that should be evaluated for multiple function device products. In addition, the examples are not intended to describe all the details of the documentation necessary to demonstrate adequate risk mitigation.
Example: Skin cancer detection software application 🔗
Product
A smart phone software application (app) that detects skin cancer from photos of suspicious lesions of moles.
Functions
Device function-under-review:
- Software app that detects skin cancer
“Other function:”
- Smart phone computing platform
- Camera on the computing platform
Impact of the “other function” on the device function-under-review
The software app depends on the smart phone camera for the photos and depends on the computing platform for the analysis.
Could there be increased risk or adverse effect of the “other function” on the device function-under-review
- The output of the camera may not be adequate for detecting skin cancer in photos of suspicious lesions of moles, resulting in misdiagnosis.
- The smart phone’s computing platform performance may not be adequate to support the software functions, including the algorithm intended to detect skin cancer in photos of suspicious lesions of moles.
Documentation in the premarket submission demonstrating that the increased risk or adverse effect that could result from the combination of functions is mitigated
- Document testing outcomes that demonstrate that there are adequate computing resources (including screen size and resolution) and error handling to accommodate the common computing platform including that the built-in camera provides adequate images.
- Document description of specific feature(s) with adequate testing outcomes that mitigate risk from software being used on a smart phone or inadequate camera.
- Documentation of specification for the use of the app with the camera and the computing platform.
Documentation of assessment in sponsor’s quality system
The sponsor should document the impact assessment and justification for their determination regarding adverse impact of the “other functions” (camera output and smart phone computing platform) in accordance with the sponsor’s quality system.
Assessment of “other function” in the premarket review
The smart phone platform and camera are not evaluated as part of the premarket review, though the testing outcomes of the device function-under-review’s performance related to using the smart phone and camera are evaluated. The software manufacturer is not responsible for electromagnetic compatibility (EMC) or electrical safety testing of the commercial smart phone.
Example: Hand-held coagulation device 🔗
Product
A hand-held coagulation (prothrombin time) test device that interfaces with a hospital information system (HIS) through a commercial off-the-shelf docking information station hardware that meets appropriate U.S. Consumer Product Safety Commission standards (electrical safety testing, safety certification, etc.) to transfer clinical in vitro test data.
Functions
Device function-under-review:
- Hand held coagulation instrument
- Coagulation (prothrombin time) test
“Other function:”
- Docking station
- Interface to transmit the data to the HIS
Impact of the “other function” on the device function-under-review
The handheld instrument depends on the docking station for charging and data transfer.
Could there be increased risk or adverse effect of the “other function” on the device function-under-review
- Charging of the handheld instrument is necessary for device performance.
- The recharging of the battery introduces new hazardous situations for the instrument itself.
- Instrument may be affected by erroneous or nontrusted data transfer from the HIS system.
Documentation in the premarket submission demonstrating that the increased risk or adverse effect that could result from the combination of functions is mitigated
- Demonstrate that appropriate mitigations were implemented to address hazards associated with battery charging.
- Document features authenticating data from trusted HIS system.
Documentation of assessment in sponsor’s quality system
The sponsor should document the impact assessment and justification for their determination regarding adverse impact of the “other functions” (docking station and interface software for data transfer) in accordance with the sponsor’s quality system.
Assessment of “other function” in the premarket review
The docking station and interface software are not evaluated as part of the premarket review, though the hazard mitigation for the docking station battery and authentication features for data transfer are evaluated as part of the device function-under-review’s safety.
Example: Traumatic brain injury determination 🔗
Product
A product that analyzes a user’s Electroencephalogram (EEG) signals recorded on a computer platform and specialized hardware and uses data generated from an electronic questionnaire to determine if the user has suffered from Traumatic Brain Injury (TBI) in an acute period after an injury.
Functions
Device function-under-review:
- Collection and recording of EEG signals
- Analyzing EEG signals and diagnosing TBI
- Presentation of results
“Other function:”
- General-purpose computing platform
- Electronic administration of questionnaire
Impact of the “other function” on the device function-under-review
The device algorithm depends on the results of the questionnaire.
Could there be increased risk or adverse effect of the “other function” on the device function-under-review
- Inaccurate input of questionnaire results may impact the performance of the software.
- Computer performance may not be adequate to support the software functions including the algorithm intended to determine if the patient has TBI.
- The output of the questionnaire may not be adequate for determining if the patient has TBI.
Documentation in the premarket submission demonstrating that the increased risk or adverse effect that could result from the combination of functions is mitigated
- Demonstrate that appropriate mitigations were implemented to limit or minimize inaccurate input to the questionnaire.
- Demonstrate that the algorithm includes features to detect inaccurate input that may impact the performance of the software.
- Testing outcomes demonstrating adequate computing resources (including screen size) and error handling to accommodate the common computing platform.
- Description of specific feature(s) with adequate testing outcomes that mitigate risk from software being used on a common computing platform.
Documentation of assessment in sponsor’s quality system
The sponsor should document the impact assessment and justification for its determination regarding adverse impact of the “other functions” (computing platform and questionnaire) in accordance with the sponsor’s quality system.
Assessment of “other function” in the premarket review
The computing platform and the questionnaire are not evaluated as part of the premarket review, though the impact to safety and performance related to using the computing platform and questionnaire on the testing outcomes and risk mitigation for the device function-under-review are evaluated. The manufacturer is not responsible for EMC or electrical safety testing of the commercial computer.
Example: Pain treatment app 🔗
Product
A Transcutaneous Electrical Nerve Stimulation (TENS) device controlled by an app on a mobile platform worn by the user to treat pain.
Functions
Device function-under-review:
- Electrical Nerve Stimulation as a treatment for pain
- App used to control the level of stimulation
“Other function:”
- Mobile platform Bluetooth transceiver and connectivity
Impact of the “other function” on the device function-under-review
The Bluetooth functionality of the mobile platform provides connectivity to the worn device enabling remote control of the stimulation.
Could there be increased risk or adverse effect of the “other function” on the device function-under-review
- The reliability and security of the Bluetooth connectivity could be compromised causing the TENS device to operate in an uncontrolled manner.
Documentation in the premarket submission demonstrating that the increased risk or adverse effect that could result from the combination of functions is mitigated
- Appropriate demonstration of applicable wireless coexistence and EMC safety standards for the intended operating environment for the TENS worn part of the device to ensure the reliability of the connection when used with the Bluetooth functionality.
- Demonstrate that appropriate cybersecurity controls are included in the design and implementation of the device function-under-review to ensure the reliability and security of the connection.
Documentation of assessment in sponsor’s quality system
The sponsor should document the impact assessment and justification for their determination regarding adverse impact of the “other function” (mobile platform Bluetooth transceiver and connectivity) in accordance with the sponsor’s quality system.
Assessment of “other function” in the premarket review
The mobile platform Bluetooth transceiver is not evaluated as part of the premarket review, though the evaluation outcomes of the device function-under-review’s safety and performance related to EMC standards and cybersecurity mitigation are evaluated. The manufacturer is not responsible for EMC or electrical safety testing of the Bluetooth product itself.
Example: Transmission of vital sign measures to an Electronic Health Record (EHR) 🔗
Product
A monitor that measures and displays vital physiological parameters and transmits them to an Electronic Health Records (EHR) system through the hospital network using a built-in Wi-Fi card.
Functions
Device function-under-review:
- Vital signs acquisition
“Other function:”
- Transmission software for sending data to the EHR system
- Wi-Fi card
Impact of the “other function” on the device function-under-review
As an integral part of the device that allows for a network connection, the Wi-Fi card has inherent risks associated with EMC and other wireless related risks (e.g., the vital physiological parameters may be corrupted in transit to the EHR system), including cybersecurity risks.
Could there be increased risk or adverse effect of the “other function” on the device function-under-review
- The Wi-Fi card may affect both the performance and safety of the device or other devices in the area.
- The network connection may affect the safety of the device by introducing cybersecurity risks.
- The misfunction of the interface software may impact the safety of the device as the vital physiological parameters may be corrupted in transit to the EHR system.
Documentation in the premarket submission demonstrating that the increased risk or adverse effect that could result from the combination of functions is mitigated
- Demonstrate that appropriate cybersecurity controls were included in the design and implementation of the product.
- Demonstrate that appropriate EMC and wireless testing was conducted and document that the Wi-Fi card does not affect the performance of the monitor.
Documentation of assessment in sponsor’s quality system
The sponsor should document the impact assessment and justification for their determination regarding adverse impact of the “other functions” (transmission software and Wi-Fi card) in accordance with the sponsor’s quality system.
Assessment of “other function” in the premarket review
The Wi-Fi card and transmission to the EHR are not evaluated as part of the premarket review, though the cybersecurity controls and performance testing of the device function-under-review are evaluated.
Example: Energy-delivering device with optional app (MDDS) 🔗
Product
Energy-delivering aesthetic device with an optional mobile application (app) that transfers treatment parameter data for cloud-based storage for later review by a physician.
Functions
Device function-under-review:
- Energy-delivering aesthetic device
“Other function:”
- Mobile app that integrates with device and transfers treatment parameter data (e.g., number of treatments, treatment parameters) to a cloud-based storage system – no real-time transmission is allowed
- Smart phone computing platform
Impact of the “other function” on the device function-under-review
The energy-delivering device function is not impacted by the mobile app or smart phone computing platform because the transmission of data cannot occur during energy delivery.
- Note: In this scenario, cybersecurity and radio frequency present risks that are inherent to the device function-under-review and are not introduced by the “other functions.” Therefore, documentation on risk mitigation for those identified risks is necessary for the premarket submission on the energy-delivering aesthetic device, and not due to the mobile app or smart phone computing platform.
Could there be increased risk or adverse effect of the “other function” on the device function-under-review
- Not applicable.
Documentation in the premarket submission demonstrating that the increased risk or adverse effect that could result from the combination of functions is mitigated
- Not applicable.
Documentation of assessment in sponsor’s quality system
The sponsor should document the impact assessment and justification for their determination regarding no impact of the “other functions” (mobile app and smart phone computing platform) in accordance with the sponsor’s quality system.
Assessment of “other function” in the premarket review
The mobile app and smartphone platform are not evaluated as part of the premarket review and no documentation on the “other functions” is necessary for premarket submission of the device function-under-review.
Example: Pulsed ultrasound and biopsy needle guide kit 🔗
Product
An ultrasound pulsed echo imaging system with software-based biopsy needle tracking functionality used to determine the depth or location of the tissue interfaces for use with biopsy needle guide kit for delivery.
Functions
Device function-under-review:
- General purpose diagnostic ultrasound system
- Biopsy needle tracking functionality
“Other function:”
- 510(k)-exempt biopsy needle guide kit
Impact of the “other function” on the device function-under-review
The biopsy needle guide kit is a convenience tray that combines a number of other ultrasound accessories that have already received 510(k) clearance (e.g., needle guide brackets, sterile ultrasound gel, sterile probe covers) or are class I 510(k)-exempt (e.g., sterile tray, drape covers). The components of the kit are intended to aid with biopsy acquisition, specifically, the biopsy needle tracking functionality that is guided via ultrasound imagery. The biopsy needle tracking functions should be compatible with the biopsy needle guide kit. There are no additional risks introduced by the biopsy needle guide kit, which has its own inherent risks, such as possibility of compromising sterility, chance of infection, or potential cross contamination, which are not introduced due to the combination with the device function-under-review.
Could there be increased risk or adverse effect of the “other function” on the device function-under-review
- The biopsy needle tracking software may not be compatible with all ultrasound needle biopsy kits. Incompatibility may lead to inaccurate or imprecise guiding of the needle to the target area.
Documentation in the premarket submission demonstrating that the increased risk or adverse effect that could result from the combination of functions is mitigated
- For biopsy needle tracking software intended to be compatible with various ultrasound needle biopsy kits, performance testing should be provided showing that the accuracy and precision meets the specifications and requirements for the subject device. For example, data can show that the software, when used with the biopsy guide kit, guides the needle to the target area within +/- 0.5 mm, which is substantially equivalent to the predicate device.
Documentation of assessment in sponsor’s quality system
The sponsor should document the impact assessment and justification for their determination regarding potential adverse impact of the “other function” (biopsy needle guide kit) in accordance with the sponsor’s quality system. The sponsor should document that any further processing of the kit and its components does not significantly affect the safety or effectiveness of any of its components in their quality system, in accordance with “Convenience Kits Interim Regulatory Guidance.”38
Assessment of “other function” in the premarket review
The biopsy needle guide kit is not evaluated as part of the premarket review, though the safety and effectiveness demonstration of the device function-under-review related to the compatibility of the biopsy needle tracking software with the biopsy needle guide kit is evaluated.
Footnotes 🔗
-
A device per section 201(h) of the FD&C Act may be comprised of one or more device functions. The term “function” is not synonymous with the term “device.” “Distinct purpose of the product,” as used in this guidance to explain the meaning of “function,” is not synonymous with “primary intended purposes” in section 201(h) of the FD&C Act. ↩
-
Available at https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfStandards/search.cfm. ↩
-
Available at https://www.fda.gov/regulatory-information/search-fda-guidance-documents/appropriate-use-voluntary-consensus-standards-premarket-submissions-medical-devices. ↩
-
Available at https://www.fda.gov/regulatory-information/search-fda-guidance-documents/standards-development-and-use-standards-regulatory-submissions-reviewed-center-biologics-evaluation. ↩
-
A software function described in section 520(o)(1)(C)-(E) of the FD&C Act is not excluded from the device definition under 201(h) if the software meets the criteria under section 513(a)(1)(C) of the FD&C Act or if the software is used in the manufacture and transfusion of blood and blood components to assist in the prevention of disease in humans (section 520(o)(4)(B) and (C) of the FD&C Act). ↩
-
Section 510 of the FD&C Act. ↩
-
Section 520(f) of the FD&C Act. ↩
-
Section 519 of the FD&C Act. ↩
-
Available at https://www.fda.gov/regulatory-information/search-fda-guidance-documents/policy-device-software-functions-and-mobile-medical-applications. ↩
-
Available at https://www.fda.gov/regulatory-information/search-fda-guidance-documents/general-wellness-policy-low-risk-devices. ↩
-
Note that a device may not be 510(k)-exempt if it exceeds the limitations outlined in 21 CFR Part 862.9 through 892.9. A similar analysis would apply for drug- or biologic-led combination products. ↩
-
“Nothing in this subsection shall be construed as limiting the authority of the [FDA] to— (A) exercise enforcement discretion as to any device subject to regulation under this Act . . .” (section 520(o)(4) of the FD&C Act). ↩
-
21 CFR 3.2(e). ↩
-
520(o)(2) of the FD&C Act. ↩
-
520(o)(2) of the FD&C Act. ↩
-
Please see the “Policy for Device Software Functions and Mobile Medical Applications” guidance document for the definition of mobile medical application, available at https://www.fda.gov/regulatory-information/search-fda-guidance-documents/policy-device-software-functions-and-mobile-medical-applications. ↩
-
“Nothing in this subsection shall be construed as limiting the authority of the [FDA] to— (A) exercise enforcement discretion as to any device subject to regulation under this Act . . .” (section 520(o)(4) of the FD&C Act). ↩
-
Please see the CDRH Premarket Review Submission Cover Sheet, available at https://www.fda.gov/medical-devices/premarket-notification-510k/content-510k. ↩
-
During an inspection, upon FDA’s request, a manufacturer must allow FDA to access, copy, and verify certain records, including the results of the design validation. See sections 704(a) & (e) of the FD&C Act; see also 21 CFR 820.180. ↩
-
One example of such a software transparency method is a Software Bill Of Materials that lists software components – including commercial, open source, off-the-shelf, and custom software components – and can aid in the management of risk in both medical device manufacturer-developed software components and purchased, third party components, which may comprise “other functions.” Please see, for example, “Healthcare Sector Coordinating Council (HSCC) Joint Strategy Plan (JSP)” Appendix G, available at https://healthsectorcouncil.org/the-joint-security-plan/. ↩
-
See 21 CFR 820.30(g) (requiring that the manufacturer establish and maintain procedures for validating the device design and that design validation shall include software validation and risk analysis, where appropriate); see also sections 704(a) & (e) of the FD&C Act; and 21 CFR 820.180 (FDA may request to review such records during an inspection). ↩
-
Available at https://www.fda.gov/regulatory-information/search-fda-guidance-documents/least-burdensome-provisions-concept-and-principles. ↩
-
We have a long-standing policy of applying the definition of indications for use in the PMA regulation at 21 CFR 814.20(b)(3)(i) in the same way in the 510(k) context. See FDA guidance “The 510(k) Program: Evaluating Substantial Equivalence in Premarket Notifications [510(k)],” available at https://www.fda.gov/regulatory-information/search-fda-guidance-documents/510k-program-evaluating-substantial-equivalence-premarket-notifications-510k. ↩
-
See, e.g., 21 CFR 801.5 (requiring that labeling include adequate directions for use, including statements of all conditions, purposes, or uses for which the device is intended (e.g., hazards, warnings, precautions, contraindications); 21 CFR 801.109(c) (for prescription devices, requiring that labeling include any relevant hazards, contraindications, side effects, and precautions under which practitioners licensed by law to administer the device can use the device safely and for the purpose for which it is intended). ↩
-
Available at https://www.fda.gov/regulatory-information/search-fda-guidance-documents/guidance-content-premarket-submissions-software-contained-medical-devices. ↩
-
Available at https://www.fda.gov/regulatory-information/search-fda-guidance-documents/guidance-content-premarket-submissions-software-contained-medical-devices. ↩
-
Available at https://www.fda.gov/regulatory-information/search-fda-guidance-documents/guidance-content-premarket-submissions-software-contained-medical-devices. ↩
-
21 CFR 807.81(a)(3). ↩
-
Available at https://www.fda.gov/regulatory-information/search-fda-guidance-documents/deciding-when-submit-510k-change-existing-device. ↩
-
Available at https://www.fda.gov/regulatory-information/search-fda-guidance-documents/deciding-when-submit-510k-software-change-existing-device. ↩
-
Available at https://www.fda.gov/regulatory-information/search-fda-guidance-documents/modifications-devices-subject-premarket-approval-pma-pma-supplement-decision-making-process. ↩
-
For more information on requirements related to combination products, cross-cutting combination product guidance documents are available at https://www.fda.gov/combination-products/guidance-regulatory-information/combination-products-guidance-documents. ↩
-
Sections 501, 502, 510, 516, 518, 519, 520(e), and 520(f) of the FD&C Act. ↩
-
“Nothing in this subsection shall be construed as limiting the authority of the [FDA] to— (A) exercise enforcement discretion as to any device subject to regulation under this Act . . .” (section 520(o)(4) of the FD&C Act). ↩
-
Available at https://www.fda.gov/regulatory-information/search-fda-guidance-documents/current-good-manufacturing-practice-requirements-combination-products. ↩
-
Available at https://www.fda.gov/combination-products/guidance-regulatory-information/postmarketing-safety-reporting-combination-products. ↩
-
Available at https://www.fda.gov/combination-products. ↩
-
Available at https://www.fda.gov/regulatory-information/search-fda-guidance-documents/convenience-kits-interim-regulatory-guidance. ↩

