
Best Practices 🔗

- Timing
- Pre-Sub’s may not be valuable if your intended use and device description aren’t set
- Otherwise, do a Pre-Sub as soon as high-risk regulatory questions are identified
- Question selection
- Focus on questions that eliminate regulatory or clinical risk. Consider that FDA allows a maximum of 4 topic (e.g., regulatory strategy, cybersecurity) categories
- Write specific questions that lead FDA to the answer you want
- Write questions that give the FDA an opportunity to disagree
- Avoid questions that FDA won’t be able to answer in a Pre-Sub or can be answered using Guidance
- Make sure you have appropriate supporting information for each of your questions. FDA will not answer questions for which they do not have sufficient information or do not understand
- Pre-Sub Preparation
- Provide detailed background information for FDA to give accurate answers. See under the Examples section below.
- Don’t provide extraneous background information. FDA reviewers are busy and have a lot of projects. If you send too much information, you’ll get less useful feedback.
- When providing background information, not only explain what approach you are using, but also explain why you are not using alternate approaches
- Use FDA terminology
- Quote FDA guidance back at the FDA
- The Call
- Plan out expected FDA questions and responses
- Don’t expect all the reviewers to have read your pre-sub carefully or to have fully understood the context
- Keep in mind that FDA Pre-Subs can be somewhat of a negotiation with FDA; there is some give and take
- Plan who will speak about which questions (we generally suggest having an regulatory consultant run the meeting)
- Don’t waste time during the meeting on unimportant topics
- If too much time is being spent on a single question with no end in sight, kindly suggest moving on to another question
- Don't be afraid to ask the FDA for their reasoning behind certain feedback
- Don't be afraid to ask for an "informal" follow up via email. FDA can be open to receiving some additional info/answering questions via email after the call
About the Author 🔗

Hi, I’m David Giese, a Partner at Innolitics. I’ve wrote this article based on my real-world experiences bringing 35+ diagnostic medical devices onto the US market. I take pride in the in-depth articles I write, and appreciate feedback and questions. I’d love to connect on LinkedIn, where I post pragmatic tips about medical-device strategy, pre-subs, and regulations for my >5k followers.
Basic 🔗
What is a Pre-Sub? 🔗
A Pre-Sub is a mechanism for requesting formal written feedback from the FDA, and (optionally) a one-hour meeting.
A Pre-Sub is appropriate when FDA’s feedback on specific questions would help guide product development, performance testing, predicate selection, or other aspects of a 510(k), De Novo, PMA, or IDE submission.
Does a Pre-Sub cost money? 🔗
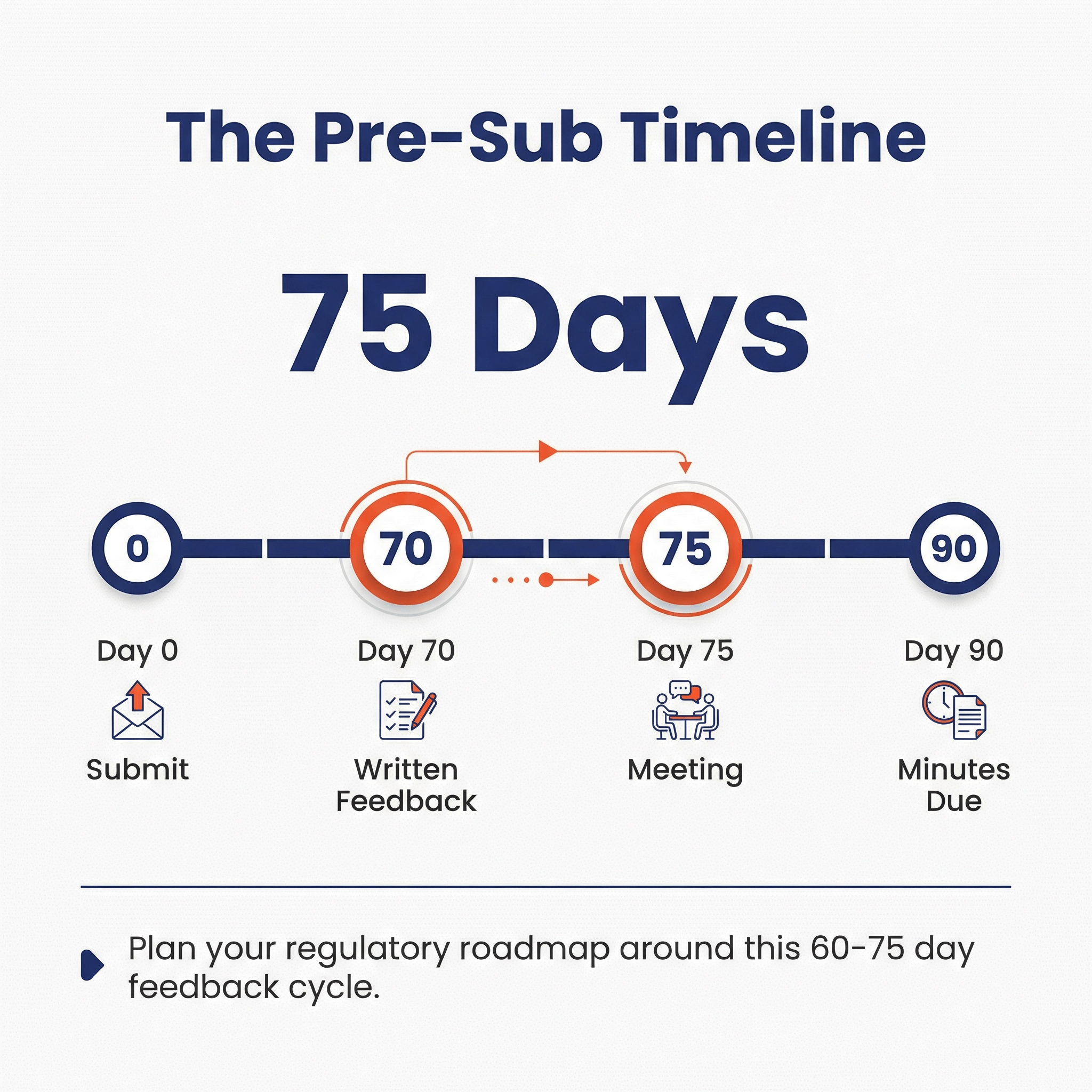
There is no FDA fee, however, preparing a Pre-Sub package takes significant effort. There is also a 60 - 75 day delay from when you submit your Pre-Sub and when you receive feedback. Time is money, as they say.
How many Pre-Subs are you allowed to do? 🔗
You can do as many as you like.
Can you do multiple Pre-Subs at once? 🔗
Although it is allowed, FDA recommends against having multiple Pre-Subs for the same device at a time, because feedback from one Pre-Sub often affects others. Therefore, unless the Pre-Subs are for widely different topics (e.g., cybersecurity and clinical study design), we generally suggest against holding multiple pre-subs at once. See this section of the guidance for more details.
Do you have to do a Pre-Sub? 🔗
No. They are always optional, but recommended in many cases.
How do Pre-Sub’s relate to 510(k)s? 🔗
Here is a diagram showing how pre-subs relate to a typical pre-market submission:
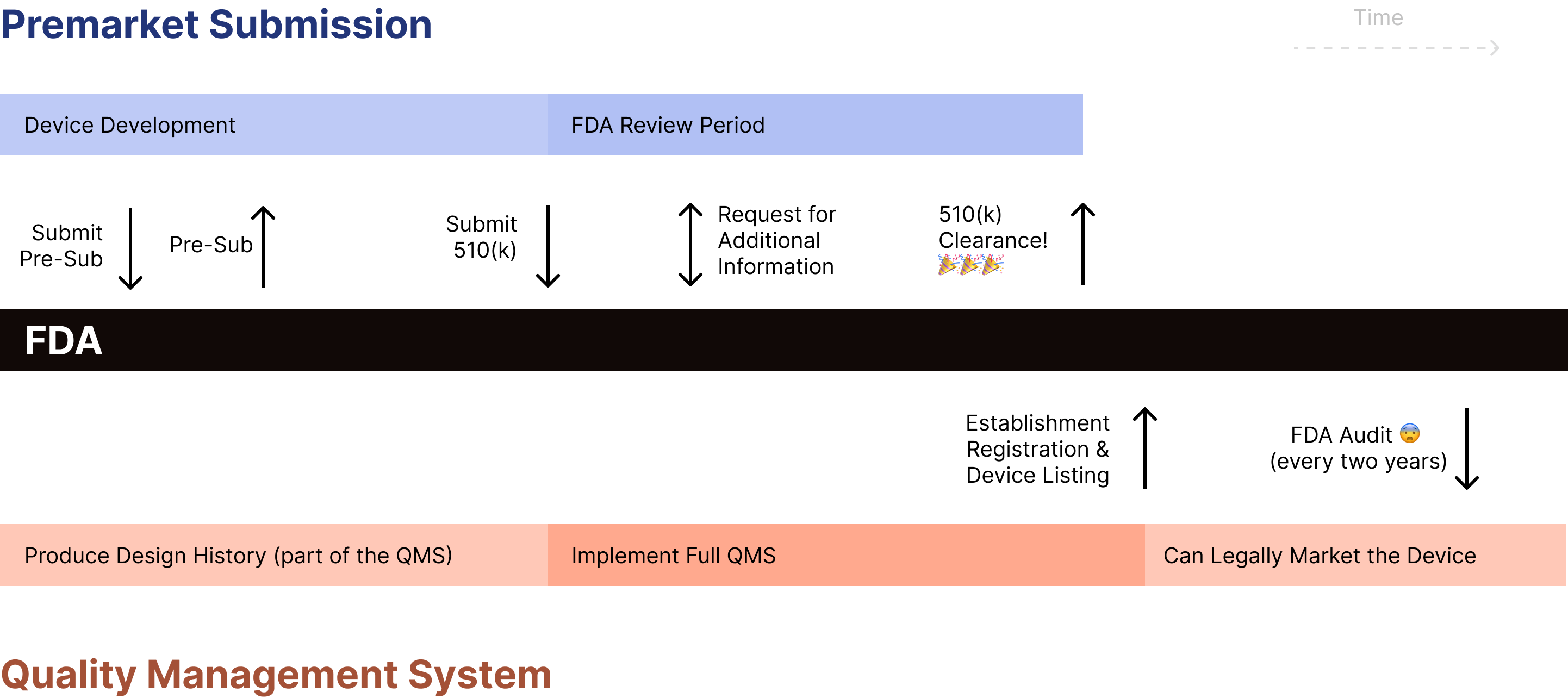
We recommend requesting a presubmission meeting with FDA early in the process (e.g., before preparing 510(k) submission, executing testing, collecting data) but once you have a clear regulatory strategy in place.
What is the process for doing a Pre-Sub? 🔗
The typical process looks like this:
- Prepare the Pre-Sub
- Identify key questions
- Write intended use, indications for use, and device description
- Write any other documentation necessary for FDA to answer questions (e.g., test protocols)
- Submit the Pre-Sub to FDA (Day 0)
- FDA provides written feedback (Day 70)
- Meeting (Day 75)
- Respond with formal minutes (By Day 90)
If no meeting is requested, then steps 4 and 5 are skipped.
The day of the meeting is mutually agreed upon with FDA, and is typically between 70 and 75 days from submitting the Pre-Sub.
Although the actual timelines for the Pre-Subs can vary and have increased and decreased over time, as of April 2024, we’re seeing that FDA is typically hitting their 60 - 75 Day goals.
Where do Pre-Sub meetings occur? 🔗
We often do them over video calls, however, FDA has started allowing in-person meetings again post COVID.
What content goes into a Pre-Sub? 🔗
Here is a summarized list of content that’s included in a Pre-Sub. Note a lot of this is quoted and slightly modified from the FDA guidance:
- Purpose. The overall purpose of the Pre-Sub including goals for the outcome of the interaction with FDA.
- Agenda. A draft agenda proposing the topics to be presented and the estimated time for each agenda item.
- Preferred Meeting Dates. Three (3) or more preferred dates and times when you are available to meet.
- Attendees. The planned attendees, including each attendee’s position, or title, and affiliation.
- FDA Experts. Any appropriate FDA staff that are requested to attend the meeting if specific expertise may be needed.
- Device Description. Listing of any relevant previous communications with FDA about the subject device. An explanation of how the device functions, the basic scientific concepts that form the basis for the device, and the significant physical and performance characteristics of the device.
- Proposed Indications for Use or Intended Use. This should include a description of the disease(s) or condition(s) the device will diagnose, treat, prevent, cure or mitigate, and a description of the patient population for which the device is intended.
- Regulatory History. Listing of any relevant previous communications with FDA about the subject device.
- Planned Follow-On Submission. FDA recommends that you clearly indicate what type of future submission (IDE, IND, CW, Accessory Classification Request, or marketing submission) is the focus of your Pre-Sub questions to help direct FDA’s feedback.
-
Background Information. FDA recommends that you include sufficient background information and supporting documents to allow them develop feedback for the Pre-Sub questions you pose. This information might include:
- literature articles
- full device description with engineering drawings
- proposed labeling
- videos
- red-lined protocol revisions
- how you addressed, or plan to address, relevant guidance documents, regulations, special controls, or other applicable sources for your device or submission type.
- Specific Questions. A Pre-Sub should include clear, specific questions to allow FDA and the submitter to focus their efforts on issues most relevant to moving a project forward. You may wish to describe your perspective on the questions you provide FDA to inform FDA’s review.
How do you prepare a Pre-Sub? 🔗
We suggest using the PreSTAR dynamic PDF to compile a Pre-Sub. You can download the PreSTAR template here. Once you download it, you can select the options for a Pre-Sub and then fill in the various sections.

Once you have filled in this information, you can see the detailed fields related to the Pre-Sub.
When is a good time to do a Pre-Sub? 🔗
It is generally too early to do a Pre-Sub if you don’t have a somewhat settled indications for use and device description. Once these are settled, we suggest doing a Pre-Sub about as early as key regulatory risks have become clear.
What are typical Pre-Sub subjects? 🔗
At Innolitics, we focus on Software as a Medical Device (SaMD) products. We most typically do Pre-Subs for the following topics:
- Clarifying a regulatory strategy or predicate-device selection
- Requesting feedback on non-clinical or clinical study plans
- PCCP design
In general, we suggest Pre-Subs to de-risk expensive medical-device development activities. Risk is higher when activities are more expensive, take a long time, or there isn’t much FDA guidance on a topic.
What is a Q-Sub? 🔗
The term “Q-Submission” or “Q-Sub” refers to the system used to track a few different types of interactions with FDA. Pre-Sub is a particular type of Q-Sub.
Informational meetings are another useful type of Q-Sub in some cases.
You can read more about other Q-Sub types here.
Where can I learn more about Pre-Subs? 🔗
The key FDA guidance is here: 2023 FDA Guidance - Requests for Feedback and Meetings for Medical Device Submissions: The Q-Submission Program
The following podcast episode with Mike Drues includes a lot of great advice for running successful Pre-Subs: June 2021 - Preparing Your Pre-Submission with the Content FDA Wants to See - Greenlight Guru Podcast.
If you are putting together a pre-sub for an AI-enabled device, I recommend my partner Yujan’s article: How to Document AI/ML Algorithms in FDA Presubmissions (Q-Sub)
Strategy 🔗
Do you always do Pre-Subs? 🔗
No, it really depends on the situation.
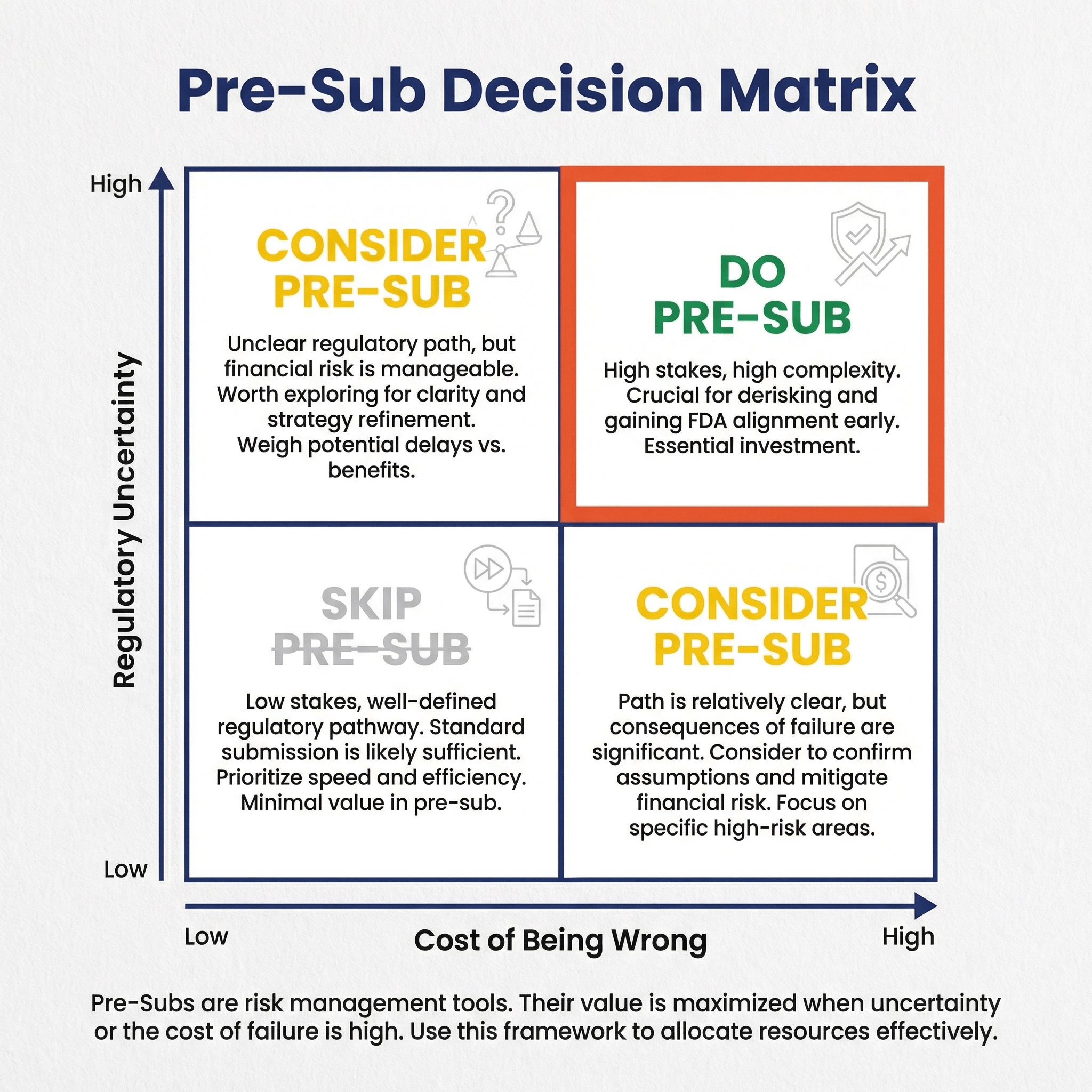
Pre-Subs are a means of controlling regulatory risk. Thus the need for a pre-sub depends on:
- The regulatory risks for your submission
- Their likelihood of occurring
- How bad it would be if they do occur (i.e., the severity)
- Other possible mitigations
Consider these two examples:
| Regulatory Risk | Likelihood | Severity | Possible Mitigations |
|---|---|---|---|
| The FDA doesn’t accept your clinical study design on a new type of device. | Moderate; it’s a new type of device without much precedent | High; re-running the study could cost $100,000+ | 1. Do a Pre-Sub |
| You’re done with development and didn’t realize you needed to do penetration testing. You think your device may not require it. The risk is the FDA requires that you do cybersecurity penetration testing. | High; FDA almost always requires penetration testing | Low; you may lose some time, but either way you’ll pay the cost of the pen testing | 1. Submit now; start pen testing in parallel; be ready with the results when FDA issues an AINN 2. Submit now; wait to see if FDA requires pen testing; if so, do it at that time (it should take less than the 180 days). |
In the first example, a Pre-Sub may avoid the need to re-run the study. In the second example, a Pre-Sub would be an unnecessary delay.
Do I need regulatory consultants to do a Pre-Sub? 🔗
You do not need a regulatory consultant to do a Pre-Sub, although it is highly recommended.
Good regulatory consultants who have experience with your type of device will have a better understanding of how FDA regulates that device type. Without them, it is very easy to waste time and money asking unnecessary questions—questions experienced consultants could answer directly.
Thus, another possible mitigation to your regulatory risks is to hire consultants with relevant experience (e.g., Innolitics is one of the world experts in SaMD and Medical Device AI). We often take over projects from other regulatory consultants after a pre-sub has gone poorly.
It is also worth noting that regulatory consultants can sometimes push to do a pre-sub when it isn’t necessary, since they can bill more for it and also it reduces the risk of them looking bad.
Regulatory consultants have experience running pre-submission meetings, and thus, can provide expert guidance on the types of questions to ask, how to ask those questions, and what type of information is needed to support them.
Are Pre-Subs a waste of time? 🔗
They certainly can be. Some common ways they are a waste of time include:
- You’re asking questions that are inappropriate for a pre-sub (e.g., you ask FDA to review data)
- You don’t provide enough context for FDA to provide concrete answers
- You don’t yet know your intended use or the desired device you want to build
Meeting 🔗
Can you request or cancel a meeting after submitting a Pre-Sub? 🔗
Requesting a meeting after initial submission: Pre-Submissions offer two options: written feedback only or written feedback followed by a meeting. You must specify whether you want a meeting in your initial Pre-Sub submission. If you request written feedback only, you cannot add a meeting later after reviewing FDA's preliminary written feedback.
Canceling a previously requested meeting: Conversely, if you originally requested a meeting but find that FDA's written feedback adequately addresses your questions, you can cancel the meeting. This saves time for both you and FDA, and allows you to move forward more quickly with your development plans. To cancel a meeting, contact the lead reviewer of the FDA review team managing your Pre-Sub. It's courteous to notify FDA as soon as possible if you wish to cancel a meeting, as this allows them to reallocate their resources.
Our recommendation: In our experience at Innolitics, we almost always recommend requesting and holding the Pre-Sub meeting, even if FDA's written feedback seems clear. The meeting provides valuable opportunities to ask clarifying questions, discuss nuances that may not be fully captured in writing, and build a rapport with the FDA review team. While there's no guarantee the same reviewers will handle your future submission, establishing a positive relationship can still be beneficial.
What are some tips for running an effective Pre-Sub meeting? 🔗
- Plan out expected FDA questions and responses
- Keep in mind that FDA Pre-Subs can be somewhat of a negotiation with FDA; there is some give and take
- Plan who will speak about which questions (we generally suggest having an regulatory consultant run the meeting)
- Don’t waste time during the meeting on unimportant topics
- If too much time is being spent on a single question with no end in sight, kindly suggest moving on to another question
- Don't be afraid to ask the FDA for their reasoning behind certain feedback
- Don't be afraid to ask for an "informal" follow up via email. FDA can be open to receiving some additional info/answering questions via email after the call
What happens if the meeting ends early? 🔗
You should never have a Pre-Sub end early!
Agree upon a few extra “informal” questions to ask FDA in the case that the meeting goes quicker than expected. If these questions are somewhat related to the existing questions, we’ve had FDA provide useful answers even if they weren’t include in the original pre-sub.
What happens if the meeting goes over? 🔗
FDA will occasionally stay on the call an extra 5-10 minutes, but don’t count on it. Typically, the call will end on the hour and if you didn’t get to some questions, well, too bad 😢.
What is the process for writing minutes for a Pre-Sub? 🔗
After the one-hour FDA meeting, you must submit minutes that FDA may review and edit.
Here are the key requirements for FDA Pre-Sub meeting minutes:
- Must be submitted within 15 days of the meeting
- Should summarize the discussion rather than being a transcript, including:
- Key discussion points
- How substantial issues were resolved
- Agreements and action items
- Should not assign statements to individuals - refer to "submitter" or "FDA" generally
- Must not include additional information or follow-up items not discussed in the meeting
- Should include any presentation slides used during the meeting
- Should include a list of all meeting attendees with their affiliations (e.g., company representatives, FDA staff). For FDA attendees, document their specific position/title at FDA. If you missed recording their exact title during the meeting, you can look up FDA staff in the HHS Employee Directory using their names.
- Submit two versions:
- Official version through DCC
- Editable version (e.g., Microsoft Word) in miscellaneous files
The FDA has 30 days to review and edit the minutes. If there are disagreements about the content, a teleconference can be scheduled to resolve the issues.
See here for additional information in the FDA guidance on the topic of minutes. Also see this example.
Minutes should be attached as an amendment to the pre-sub.
How should meeting minutes be formatted? 🔗
Meeting minutes should be written in complete sentences using a clear paragraph format. The FDA guidance states that minutes should "summarize the discussion" rather than being a transcript. While the need to summarize discussions may lead some people to gravitate towards using bullet points, but we believe complete statements and paragraphs are more formal and appropriate for official FDA documentation. Writing in complete sentences helps ensure clarity and professionalism in the official record. The minutes don't need to be overly formal or verbose, but using full sentences and paragraph format demonstrates the formality expected for documents that become part of your official regulatory file.
Meeting minutes should include a preamble that provides key information about the meeting:
- Meeting type (e.g., "Pre-Submission Meeting")
- Q-Sub number
- Meeting date and time
- Meeting format (e.g., virtual via Microsoft Teams, in-person at FDA facility)
- Complete list of attendees with their affiliations (for FDA staff, include their specific titles/positions)
- Device overview and intended use (summary)
Following the preamble, organize the minutes by question, with each section including the original question as submitted and a summary of the discussion and any agreements reached. Additionally, any discussion items that came up during the meeting but were not part of the original pre-sub questions should also be documented in the minutes.
Why are the Pre-Sub minutes important? 🔗
The minutes are crucial because they become part of your official file and will be referenced by FDA reviewers during future submissions. When FDA agrees to a specific testing approach and confirms it in the minutes, they are less likely to change their position later (though this remains possible).
For this reason, be sure to document all decisions made and agreements reached in the meeting minutes. We suggest putting the most important ones in a list at the top to be sure the reviewers will read them.
Examples 🔗
Additional Data Collection Situation 🔗
Pre-subs require time and money to prepare, and they add a 60-70 day delay to your roadmap. Therefore, they only make sense when they help de-risk a decision whose cost significantly outweighs the cost of the pre-sub itself. Here's an example to illustrate this:
Consider an AI-enabled device where product development is complete and you're ready to submit. You know the FDA may request a subgroup analysis of your model's performance across different ethnic groups. However, you have several academic papers suggesting that ethnicity won't affect your device's input variability. Should you:
- Do a pre-sub to ask FDA about collecting the data and proceed based on their response?
- Submit the 510(k) as-is and address FDA's feedback if needed?
The answer depends on the time and costs of collecting the data. If data collection and analysis is relatively quick (say, 2 months), then option (2) makes more sense. If FDA disagrees with your justification for not including ethnic data, you can collect it within the 180-day hold clock.
However, if you're early in product development and the pre-sub won't delay your submission timeline, it's worth doing the pre-sub to de-risk the decision and avoid lengthy holds during review.
Questions for De Novo Pre-Subs 🔗
The FDA’s 2021 De Novo Classification Guidance includes some sample questions for pre-subs before a De Novo. They also mention special information that should be included if you’re doing a De Novo.
Examples of questions to pose to FDA in a De Novo Pre-Sub include:
- Based on the device description, its intended use/indications for use, and/or technological characteristics, and information on the search performed for legally marketed devices, does FDA believe the device is eligible for De Novo classification?
- Are there other risks, in addition to those identified in the Pre-Sub, given the indications for use for the device?
- If applicable, are there controls that should be considered to provide a reasonable assurance of safety and effectiveness for the device?
- Are the non-clinical study protocols sufficient to allow the collection of data from which conclusions about device safety and/or effectiveness can be drawn? For example:
- Is the identified level of concern the appropriate level of concern for the device software?
- What, if any, additional biocompatibility and/or sterility testing would be appropriate?
- If clinical data are needed, are the proposed study design and selected control group appropriate?
Bad Example #1: Too Many Algorithms 🔗
Background 🔗
In a past presubmission meeting, we presented a device with multiple AI algorithms, each targeting different anatomical areas and providing distinct outputs. Our aim was to maximize the meeting's value by seeking FDA feedback on as many algorithms as possible. However, upon receiving the written feedback, we realized this approach had backfired. We had overwhelmed the FDA with information, obscuring the device's overall intended purpose. Consequently, the FDA couldn't address many of our original questions. This misstep forced us to spend a significant portion of the meeting clarifying our device description, workflow, and intended use. Even then, the FDA couldn't offer final recommendations during the call due to their initial lack of understanding about the device.
Pre-Sub Excerpt 🔗
Sponsor Question
Does the FDA have any concerns about the proposed marketing claims and the methods for supporting them?
Official FDA Response
We are not able to answer this question before we fully understand the intended use of your device. Please see the FDA’s response to Question 1, which discusses our current thinking of the functionalities and indications of the device, the choice of predicates, and regulatory strategy currently proposed.
CLIIP Background Information for SaMD 🔗
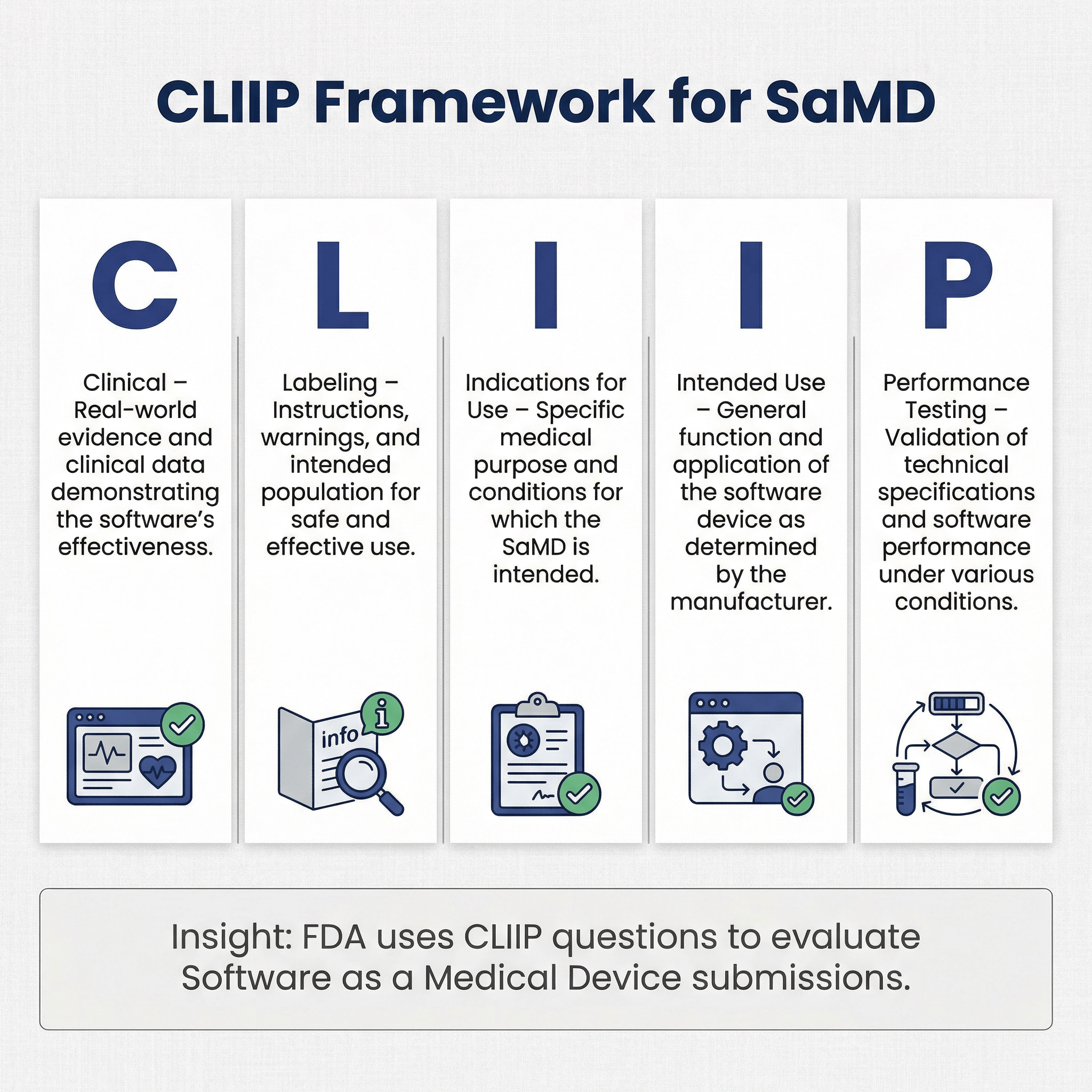
In one of our recent presubmissions, FDA shared a "CLIIP Questions for Software Review" document with us. CLIIP stands for “clinical, labeling, indications for use, intended use, and performance testing”. This document is a way for the FDA to share informal recommendations to provide some guidance for all the information for a SaMD that may be required for a review of the device.
Below is a copy of the CLIIP questions.
-
Please ensure that your device description addresses as many of the following questions and
considerations, thinking of the clinical context of your software and how that relates to the intended use, software environment, and the analysis capability of your software.-
Software Environment (Inputs and Outputs):
- What are the major inputs (data, images, measurements, sensors/attachments etc…), and who provides these inputs? What is the format (e.g. questionnaire, image, report, etc…) of the inputs? Are there any specifications that must be met for the SaMD to work with associated hardware? What are the primary outputs, and to whom are they provided? What is the format of the output, and what is the end user or patient expected to do with these outputs?
- What is the data or information flow of your device – are inputs provided or outputs delivered locally, via cloud, by disk or drive, wirelessly/RF etc…?
- Does, or how does, your software interface with a networked device, any off‐the‐shelf software (medical or non‐medical), cloud or network storage, etc…?
-
Analysis:
- Does your software perform an analysis? Please describe the analysis environment [simple rule-based calculations, online administering of a test, AIML, neural network, fixed or adaptive algorithm].
- Does it replace any part of what a physician does (e.g. automates steps, triages patients, provides a definite diagnosis or recommends/performs treatment), or does it perform clinical decision support functions? For example, triages priority cases for physician review, identifies region of interest for further review, suggests likely diagnosis but requires confirmation by physician, recommends treatment plan?
-
Clinical Context:
- What is the intended use of the software device?
- Who operates the software, and who is the patient? Is the user the patient, a caregiver, or a healthcare professional? Does the software focus on a specific disease, condition, or patient characteristic/demographic? Does the software provide information that is directly applicable to a specific disease or condition? How does your software change or support current clinical workflow?
-
Software Environment (Inputs and Outputs):
-
How does your proposed testing plan align with how your software is intended to be used?
-
What clinical evidence have you acquired to demonstrate that clinical performance goals are met?
- What published literature or other real‐world evidence exists that supports your
software’s clinical use? - What modeling/simulation or other alternative methods did you use in lieu of clinical
testing to support your software’s clinical use? - How do the results of the clinical evidence discussed above align with your software’s intended use and design (#1 above)
- What published literature or other real‐world evidence exists that supports your
-
How does your proposed non‐clinical testing plan and validation align with your high level
design requirements? With identified hazards/vulnerabilities?-
What bench testing or non‐clinical validation was performed to meet device
specifications / high level design requirements? -
What testing or validation that was performed to address your device’s most significant hazards (identified above) and to mitigate associated risks?
What testing was performed to ensure that interfacing hardware (sensors, accessories, other systems) successfully integrates with the SaMD? -
What testing was performed to ensure that the SaMD communicates with other
components of the system (e.g. SaMD on one mobile platform communicating with
SaMD on another platform)? This includes software environment testing for networked devices.
-
What bench testing or non‐clinical validation was performed to meet device
-
What clinical evidence have you acquired to demonstrate that clinical performance goals are met?
-
How have you considered and addressed your software’s most significant risks or vulnerabilities through your Quality system? Specifically, are there and how have you considered and addressed:
- Device‐specific hazards that could change the risk categorization of your software (by
changing either a) the state of health care situation or condition, or b) the significance of
information provided by the SaMD to the health care decision)? - Hazards unique to your software based on its clinical or medical use (relating to missed or inappropriate diagnosis, incorrect or delayed treatment, corruption or lost patient data,
adaptive algorithm impacting clinical intended use or context or introduce new risks, etc..?) - Failures distinct to your software’s use environment (mobile, cloud, short range RF, etc…?)
- Hazards of misuse arising from labeling?
- Device‐specific hazards that could change the risk categorization of your software (by
-
How does your software’s labeling align with its intended use and the results of your performance testing?
- How does your labeling explain or summarize the performance of the software (clinical and/or performance testing endpoints) and the safety and effectiveness of the software?
-
Depending upon the certainty of the output, are there situations where your software could cross the line between different IMDRF categorizations (informing/driving clinical management/ diagnosing or treating)? If so, how does your labeling clearly identify when such situations could happen and that your software is performing within its intended use?
-
If applicable, does your labeling clearly identify how your device outputs could
potentially alter clinical workflow? How does your labeling ensure that the end user will operate to correctly apply outputs according to its intended use?
-
If applicable, does your labeling clearly identify how your device outputs could
- What information or training will the end user need to have to operate the software as intended (including how to ensure that the inputs are appropriate and that the outputs are used correctly)?
-
Are you considering making any claims in the future (in labeling or promotional literature) such as:
- Claims outside the current stated intended use
- Claims not currently supported by clinical or non clinical testing
-
Claims that change the clinical context of the software (e.g. relating to improved
treatment, reduced side effects, diagnosis of a disease, etc…)
Revision History 🔗
| Date | Changes |
|---|---|
| 2024-04-23 | Initial Version |
| 2024-08-22 | Added a couple of examples, including the CLIPP questions. Clarified a few of our earlier points. |
| 2024-12-17 | Added another example of a situation when you shouldn’t do a pre-sub. Added another section with questions and answers related to running the meetings. Expanded the answers to some existing questions. |
| 2025-11-10 | Added more questions about the strategy of doing a pre-sub, when you’d do one vs not do one, and so on. Add example questions from the De Novo guidance. |



