

Introduction 🔗
The FDA says developing your design inputs is “the single most important design control activity,” yet writing good design inputs is surprisingly difficult. This article presents Innolitics’ answers to questions our clients frequently ask us about design inputs. It also analyzes a variety of poorly written example requirements.
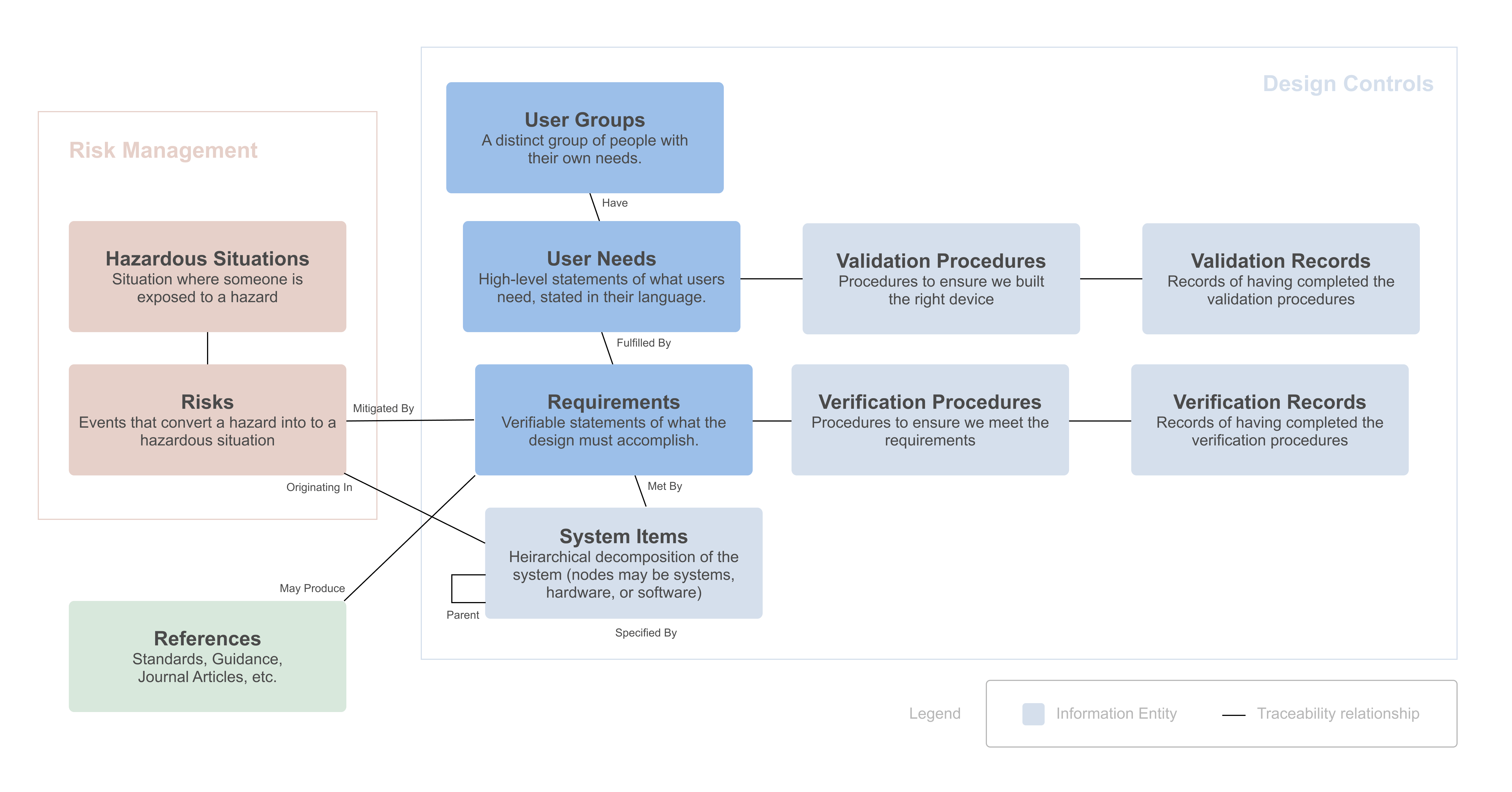
The following diagram shows how requirements and user needs related to other information entities that are collected when developing a medical device:
Innolitics specializes in Software as a Medical Device (SaMD). Many of the examples are software-related, but the principles apply to all medical devices.
Reach out if you have questions or comments. If you need help writing or verifying your design inputs, or if you’re looking to outsource your medical device software development or regulatory affairs activities, we’ll be happy to help get your device on the market as quickly as possible.
If you like this article, you may also be interested in our other “FAQ, Best Practices, and Examples” style articles:
- Medical-Device Software Regulations: Best Practices, FAQs, and Examples
- Design Inputs: Best Practices, FAQs, and Examples
- SBOMs: Best Practices, FAQs, and Examples
We are a software development and regulatory consulting firm that specializes in SaMD. We help manufacturers with cybersecurity, regulatory strategy and submissions, and provide end-to-end SaMD development.
About the Author 🔗

Hi, I’m David Giese, a Partner at Innolitics. I’ve wrote this article based on my real-world experiences bringing 35+ diagnostic medical devices onto the US market. I take pride in the in-depth articles I write, and appreciate feedback and questions. I’d love to connect on LinkedIn, where I post pragmatic tips about medical-device software, AI, and cybersecurity for >5k followers.
Basics 🔗
What is a requirement? 🔗
A requirement is a statement about something the medical device must accomplish.
Good requirements are
- necessary — you can’t take it away
- irreducible — it can’t be split into smaller requirements
- not prescriptive — it can be met using multiple different designs
- verifiable — it can be written in a way that you can check that it’s met.
What is a design input? 🔗
The regulations and standards aren’t especially clear about distinguishing these two terms.
In practice, most design inputs are requirements, and thus the terms are used interchangeably.
If you want to make a distinction, then we’d say a design input is any information, including requirements, that the design team uses as an input when performing their medical device design process. The results of the design process are design outputs.
What is a user need? 🔗
User needs are general statements of, well, what your users need! They should be expressed in terms that your users would say or how you would write marketing material to attract these users. They tend to be more abstract than requirements.
Good user needs are
- real — they originate from actual users!
- goal-oriented — describe what goal or outcome the user needs to achieve
- not technical — doesn’t use technical jargon that the user wouldn’t use themselves
- validatable — it would make sense to ask users if you met the need.
What is a user group? 🔗
A user group is a distinct group of users with different needs. They will typically interact with your device in different ways. E.g., a radiologist may use your device to produce a report while the oncologist will read the report.
The Design Controls Guidance discusses this in detail:
Particular care should be taken to distinguish among customers, users, and patients to ensure that validation addresses the needs of all relevant parties. For a consumer device, the customer, user, and patient may all be the same person. At the other extreme, the person who buys the device may be different from the person who routinely uses it on patients in a clinical setting. Hospital administrators, biomedical engineers, health insurance underwriters, physicians, nurses, medical technicians, and patients have distinct and sometimes competing needs with respect to a device design.
How do requirements and user needs differ? 🔗
Requirements need to be precise enough to be objectively verifiable, while user needs can be more subjective. A user may say “I need the software to be easy to use.” That’s not very precise, but it is what they need, and you can subjectively validate that the software meets the need by getting five users to try the software and confirm they find it “easy to use”.
Of course, the user needs should be as clear and unambiguous as you can make them. So if a user says they want it to be “easy to use”, you can probe further into what they mean. Does “easy” mean similar to existing user interfaces? Does it mean it is integrated into existing systems?
Also, unlike requirements, which shouldn’t contradict, user needs can be in a little bit contradictory. After all, the needs of one user group are in tension with the needs of another.
If you can’t express a user need in language that would make sense to a user group, then it might actually be a requirement. You can get back to the user need by asking, “Why is this a requirement?” Usually this question takes you up a level of abstraction, such that you can then identify the generic user need that that requirement is meeting.
How do system requirements and software requirements differ? 🔗
If your device only contains software, then your software requirements may be the system requirements.
For hardware and software devices (or complex software-only devices), it’s useful to organize your requirements into a hierarchy of increasingly specific requirements. System requirements, aka product requirements, are the top-level requirements. The next level of requirements are typically split into various subsystems, each with their own requirements. Thus, the software subsystems have software requirements.
System requirements are often presented in a System Requirements Specification (SyRS) or Product Requirements Document (PRD). The lower-level requirements are often written by disparate team members and are presented to the FDA in separate documents. Software requirements are presented in a Software Requirements Specification (SRS). Hardware subsystems can be presented separately, too.
Traceability 🔗
Why trace user groups to user needs? 🔗
Different user groups may have different needs.
If you see contradictory needs, it may be because you have different user groups.
If you have two user groups that all have the same needs, you may want to consider simplifying and combining them.
Why trace requirements to user needs? 🔗
Tracing requirements to user needs helps identify undocumented requirements and needs. When a requirement can't be traced to any existing user need, you might have an undocumented need—or perhaps the requirement is unnecessary.
This traceability also provides valuable design history for future engineers who may wonder why the device was designed a certain way when considering changes. As a software engineer, I've often refactored designs only to later realize there was a good reason for the original approach.
Furthermore, this traceability is required by the Design Controls Guidance.
Here are a few observations about tracing requirements to user needs:
- Typically, there are many more requirements than user needs.
- While writing requirements, you might discover that some user needs should be requirements.
- Risk-control requirements often don’t originate in a vocalized user need, because users aren’t aware of the technical details of the device. I will therefore trace risk control requirements to a generic user need along the lines of “The patient needs the device to be safe”.
Why trace risks to requirements? 🔗
When particular risks are mitigated by the design of the device (i.e., made less likelier to occur or less severe when they do), then you want to create requirements for this part of the design, thus ensuring that the mitigation works.
Tracing requirements to risks preserves a record of the mitigations that are in place. It also keeps a record of why certain requirements are present so you know whether its safe to remove or change them.
This traceability is required by section 4.5 of ISO 14971.
Can a requirement trace to multiple user needs? 🔗
Yes, although typical requirements trace to just one user need, occasionally a requirement is necessary to fulfill multiple user needs. If a requirement traces to too many user needs, it becomes less clear which need is the most important.
Why trace requirements to software items? 🔗
This ensures that each requirement has been fully implemented.
This traceability is required by IEC 62304.
Why trace requirements to verification protocols? 🔗
To ensure that the requirements are met and the device is thus acting as expected.
Writing User Needs 🔗
How do you collect user needs? 🔗
The best way is to actually talk to users!
Building a medical device is a long and arduous road. It is tragic how often people will climb over the product development and FDA mountains, only to realize their is not a product-market fit. I.e., you didn’t know what your users actually needed! Thus, we encourage our clients to spend a lot of time early on talking to real users about their needs. It can feel like a waste of time, but in our experience, it always pays off.
What format should we use when writing user needs? 🔗
We typically write them like this: “The physicians need the software to not disrupt their workflow.”
In this example, “physicians” is the user group.
What’s the difference between a business need and a user need? 🔗
Sometimes requirements for a device originate from business needs that are distinct from what your user needs. For example, a business need may be “To minimize the cost of the device.” In general, the FDA is not interested in business needs or the device requirements that originate from them, so it is best to exclude them from the documentation you provide the FDA to minimize the opportunity for reviewers to ask questions about these topics.
That said, from a workflow standpoint, it is convenient to track business needs and their downstream requirements within the same tool. Our Medtech OS supports this functionality.
How do you check if your user needs are good? 🔗
First, individually look at each user need and ask whether it’s:
- real?
- goal-oriented?
- not technical?
- validatable?
Examples of these are presented at the end of this article.
You can test if a user need is real by ensuring it came from or was checked by a real user who is a member of that user group.
You can test if a user need is goal-oriented by reading it and asking if it’s tied to a goal or desire of the user group’s members.
You can test if a user need is non technical requirements by checking if a user would state it in their own words. If it includes technical lingo that a user wouldn’t know about, it’s likely a requirement.
You can test if a validatable if it would make sense to ask a member of that user group if the need was met.
Second, you need to consider the user needs collectively:
- Can any user needs be combined?
- Do you cover the needs of all user groups?
- Do you cover the business needs?
- Are you missing any user needs?
Writing Requirements 🔗
How should requirements be written? 🔗
Typically requirements follow the form “THING must HAVE PROPERTY” or “UNDER CONDITION, THING must HAVE PROPERTY.”
Conventionally, the word “shall” has been used to denote requirements. However, we suggest using the word “must” instead of “shall.” “Shall” is ambiguous, and rarely occurs in everyday conversation. The legal community is moving to a strong preference for “must” as the clearest way to express a requirement or obligation. So states the Plain Writing Act of 2010.
(For a good discussion of “shall” and “must”, see lawyer, lexicographer, and teacher Bryan Garner’s A Dictionary of Modern Legal Usage (2d ed. 1995), pages 939-942.)
How big should individual requirements be? 🔗
Requirements need to be stated in terms that permit the establishment of test criteria and performance of tests to determine whether the criteria have been met. Thus, it’s usually convenient to use small statements, each having a unique identifier that can be traced to a verification protocol. There is no regulatory reason why you couldn’t include several statements under a single identifier, but doing so will make verification more error-prone.
For example, if REQ-10 is a few paragraphs long, how will you know every item it discusses has been verified? It’s often better to split out a big requirement into smaller parts that can be verified individually. For example, you could split REQ-10 into REQ-10.1, REQ-10.2, ... , REQ-10.15. The traceability analysis would ensure you verify all 15 parts.
Why should requirements avoid prescribing a design? 🔗
There are a few reasons why requirements should say what is required and not how each should be met.
First, you’ll need a lot more requirements; these will make your verification more expensive. Also, a lot of verification steps won’t be especially useful since you’ll be checking secondary design choices rather than fundamental requirements the design must meet.
Second, it’ll be more costly to update the software if the desired design changes. Your requirements and verification protocols will be tied to a particular design, rather than to the requirements that design must fulfill.
For example, you could have a requirement that says:
REQ-1: The software must use Yocto to generate its operating system image.
This requirement could be verified by inspection, but it’s a bad requirement for a couple of reasons. It specifies using a particular tool, Yocto, to generate the OS image. Why Yocto? Probably because we want the images to be small and for the device to boot quickly.
But it would be better to replace this requirement with:
REQ-2: The software must boot in less than 30 seconds.
REQ-3: The OS image must be smaller than 200 MB.
These verifications will be easier and more useful. Note that REQ-1 could be met while REQ-2 and REQ-3 are not. And you’ll need to update REQ-1 if you ever switch from Yocto to a better tool. Had you used REQ-2 and REQ-3, you wouldn’t need to update your requirements or your verification protocols.
All of that being said, there are situations when you may want to prescribe a design. The 1997 Document Controls Guidance has a nice comment about this:
There are occasions when it may be appropriate to specify part of the design solution in the … requirements. For example, a manufacturer may want to share components or manufacturing processes across a family of products in order to realize economies of scale, or simply to help establish a corporate identity. In the case of a product upgrade, there may be clear consensus regarding the features to be retained. However, it is important to realize that every such design constraint reduces implementation flexibility
How many requirements do we need? 🔗
It’s possible to err by having too few or too many. Here are a few heuristics:
- Add requirements for anything that needs to be verified.
- Be more comprehensive in the high-risk parts of your device.
- Requirements should be useful inputs for engineers who will produce the design.
- Don’t use requirements to describe how the device works, but rather what function it provides.
Sometimes, especially if you’re writing them for software that’s already been built or for which a design has already been selected, it’s easy to write requirements to prescribe a particular design. Good requirements don’t describe the design; rather, they describe what the design needs to accomplish to be successful.
Occasionally there might be a business reason why a particular design must be used. For example, you must use the Python programming language since many people on the engineering team know Python already. In this case, it makes sense to “pin part of the design down” with requirements instead of letting engineers dream up their own design. However, these sorts of requirements shouldn’t be used if they aren’t, well, required.
How should requirements be organized? 🔗
There are a variety of ways to organize software requirements. The best way depends on the tools you’re using.
Regardless of the tool you use to collect and trace your requirements as part of your development, the Software Requirements Specification (SRS) that you provide to the FDA must be “well-organized, easily navigable, and readable with the labeling and/or grouping of requirements by function” (per the 2023 Software Guidance).
Do you recommend any tools for handling traceability of requirements? 🔗
This is a large topic that we hope to write about more in the future.
We like using Notion to track our various related regulatory information items.
Why does each requirement need a unique ID? 🔗
Requirements will be listed in documents submitted to the FDA. A unique ID is needed so each requirement can be referenced from other documents (including, possibly, FDA reviewer correspondence).
Can I use GitHub Issues (or Jira Issues) as requirements? 🔗
The 2023 Software Guidance says:
FDA acknowledges that modern development practices may employ incremental or evolutionary software development practices. Additional forms of software requirements might be included in the submission, such as well elaborated stories, use cases, textual descriptions, screen mockups, and data flows.
Thus, the FDA is likely OK with you using GitHub issues so long as each one is uniquely identifiable, unambiguous, verifiable, and doesn’t contradict other issues. However, we generally recommend against using user stories to track your requirements.
Doing so introduces a few difficulties. First, you would need a way to address changing user stories. Say you just use a simple issue tracking system and close out issues once they’re complete. Now if requirements in Issue 403 are partially overridden by Issue 892, which are the correct requirements to include in the next regulatory submission? This can get difficult. Issue trackers tend to track changes, while the SRS needs to be a complete, unambiguous, and non-contradictory list of current requirements.
How do user interface mockups relate to software requirements? 🔗
The 2023 Software Guidance says:
FDA acknowledges that modern development practices may employ incremental or evolutionary software development practices. Additional forms of software requirements might be included in the submission, such as well elaborated stories, use cases, textual descriptions, screen mockups, and data flows.
Thus, it’s fine to include user interface mockups with your other requirements.
Usually your software won’t look exactly like the user interface mockups. Thus, if mockups are included in your requirements, corresponding verification protocols must indicate how much the software can vary from the mockups.
Sometimes, if the precise design of the mockups isn’t really “required,” it might be better to treat UI mockups as if they’re design outputs rather than design inputs. Both approaches are valid; it really depends on whether it’s important to verify that the UI matches a particular design or if the suggested UI isn’t that important.
How do you verify your requirements are good? 🔗
First, individually look at each requirement and ask whether it’s:
- verifiable?
- necessary?
- overly prescriptive?
- too large?
Examples of these are presented at the end of this article.
You can test if a requirement is verifiable by writing a verification protocol. Ambiguous, unrealistic, or negative requirements won’t be verifiable.
You can test if it’s necessary by asking: “What is the worst that could happen if we take this away?”
You can identify overly prescriptive requirements by asking why it’s stated the way it is. If another design would be OK, then you probably need to move the requirement up a level.
You can assess if a requirement is too large by determining if you can split it up into smaller, independently verifiable parts.
Second, you need to consider the requirements collectively:
- Do any requirements contradict each other?
- Do any duplicate one another?
- Are you missing any requirements?
How do you identify missing requirements? 🔗
Identifying missing requirements is often more difficult than identifying issues with an existing requirement. Here are a few sources of inspiration when identifying missing requirements:
User manual and labeling – Are all of the major functions specified in your user manual (or other labeling) covered by requirements?
User needs – Does each of your user needs have an associated system requirement? Can you think of related functionality associated with a user need that isn’t covered? (A similar analysis can be done when tracing software requirements to system requirements.)
Design outputs – Although it’s beneficial to try to write your design inputs before performing the design procedure that produces design outputs, the reality is that design is a cyclical process.
As you begin working you’ll identify new design inputs. Thus, you can identify missing requirements by looking through your design (or a prototype) and looking for areas that don’t have originating requirements.
Risk Management – Risk control measures should trace to requirements. You should probably search the MAUDE database and similar sources (i.e., Total Product Life Cycle) to identify adverse events that have occurred in your device’s product code. Such events can be used to understand the types of risks your device might have.
Past products or competitor products – Look through past projects or competitors’ websites.
Standards and guidance documents – The FDA publishes a variety of guidance documents for differing device types. These documents often include requirements, written by experts, that you might not have thought of on your own. Standards might play a similar role.
What standard or guidance documents should we refer to? 🔗
There may be guidance documents that are relevant for your particular device, but here are a few that commonly apply:
The Software Validation Guidance provides a list of software requirement areas to cover.
The IEC 62304 standard requires you to cover these areas:
- functional and capability requirements
- performance (e.g., purpose of software, timing requirements),
- physical characteristics (e.g., code language, platform, operating system),
- computing environment (e.g., hardware, memory size, processing unit, time zone, network infrastructure) under which the software is to perform, and
- need for compatibility with upgrades or multiple SOUP or other device versions.
- Software system inputs and outputs
- data characteristics (e.g., numerical, alphanumeric, format)
- ranges,
- limits, and
- defaults.
- interfaces between the software system and other systems
- software-driven alarms, warnings, and operator messages
- security requirements
- those related to the compromise of sensitive information,
- authentication,
- authorization,
- audit trail, and
- communication integrity,
- system security/malware protection.
- user interface requirements implemented by software
- support for manual operations,
- human-equipment interactions,
- constraints on personnel, and
- areas needing concentrated human attention.
- data definition and database requirements
- form;
- fit;
- function.
- installation and acceptance requirements of the delivered medical device software at the operation and maintenance site or sites
- requirements related to methods of operation and maintenance
- requirements related to IT-network aspects:
- networked alarms, warnings, and operator messages;
- network protocols;
- handling of unavailability of network services.
- user maintenance requirements
- regulatory requirements.
The 2025 Cybersecurity Guidance includes a list of cybersecurity controls, many of which will result in software requirements. The MDS2 form is also a good source of cybersecurity requirements.
Annex C of AAMI TIR57:2016, Principles for medical device security—Risk management, also has some good questions for generating cybersecurity-related requirements.
Who should write software requirements? 🔗
Often business analysts or project managers (PMs) will write the system requirements. It can be difficult, however, for non-engineers to write the lower level requirements.
Since engineers are often very involved with design and verification steps, it’s helpful for them to be able to write requirements.
That being said, gathering and documenting software requirements requires a different skill than software development and in our experience, many engineers aren’t interested in learning how to do it well. Also, it can be difficult for engineers to avoid prescribing a design with the requirements since, after all, they’re usually thinking at the design-level.
When should you write requirements? 🔗
Attempting to comprehensively document all requirements before starting product development is a mistake. This was a key insight of the agile movement that is particularly true for SaMD, since software can be changed more easily and quickly than hardware.
It is also a mistake to not document requirements as you go, since all software projects have requirements—whether you attempt to write them down or not.
I informally start to write requirements early in our projects, sometimes even during the early sales conversations. We then will try to document requirements before implementing the corresponding software, since when you write out the requirements, it helps you identify unknowns and hidden assumptions. Written requirements also help align the team.
Throughout the project, we continue adding new requirements, especially during safety and security risk assessments.
You should avoid waiting to document the requirements until after the software is built, since the activity is less useful when done that late.
We stop adding requirements during the design review just before verification and validation.
Can you recommend other resources on this subject? 🔗
Yes! Ivy Hook’s concise article, “Writing Good Requirements”, is quite useful.
We also suggest reading chapter 21 of David A. Vogel’s, Medical Device Software: Verification, Validation, and Compliance.
Finally, although it’s not written for medical devices and it can be long-winded and fluffy in places, Suzanne Robertson and James Robertson’s book, Mastering the Requirements Process, 3rd Edition, has a lot of useful content.
Examples 🔗
Bad User Needs 🔗
The device needs to automatically reconnect to the mobile phone upon disconnection.
This user need is focused on the device, and not the user. Why would a user want the device to automatically reconnect? After considering this, you may arrive at something like this: “The patients need the device to work seamlessly, even after losing a connection.”
The patients need the device to minimize their cognitive load.
A typical patient is unlikely to state this need using the phase “minimize their cognitive load.” E.g., in your design validation, would you ask a patient “Did the device’s design minimize your cognitive load while using it?” Instead, you may want to state it like this: “The patients need to be able to use the device while multi-tasking.”
Unclear Requirements 🔗
The user shall be able to take a screen capture of any of the screen within the software.
As written, this sentence isn’t explicitly stating a requirement of the software. Instead, it is making a statement about what the user must be able to do. What if the user can take a screenshot using the operating system. Would that suffice or does the software need to provide a screenshot mechanism? As written, this is ambiguous. A better way to write this would be: “The software shall provide a mechanism for users to capture a screenshot of any screen.”
Unverifiable Requirements 🔗
The system must allow the user to install upgrades, depending on user role.
This couldn’t be verified unless one knew which user roles can install upgrades. Thus, it should be rewritten: “The software must allow ‘Admin’ and ‘IT’ users to install upgrades.”
The viewer must support various DICOM modalities, such as CR, DX, CT, MR, US, and PET.
This requirement isn’t verifiable since it’s not clear what is included in “various DICOM modalities.” It should be updated to list the full set of modalities that are supported. The verification protocol would likely need to check each modality in some fashion.
The system must support multi-touch gestures on all pages.
Unless “multi-touch gestures” is defined more precisely somewhere else in the SRS, this requirement is too vague to be verified. Which multi-touch gestures need to be supported? What will they do? It needs to be split into smaller, verifiable requirements or “multi-touch gestures” needs to get a precise definition.
The system must be able to send email messages to users.
Without knowing when emails should be sent to users, it's not clear how you would verify this requirement. This can be solved by making the requirement more specific: "The software must send emails to the ordering physician when the test completes.”
The system must be developed to be in compliance with FDA guidance requirements.
The FDA has a lot of guidance documents. Some, such as its “General Principles of Software Validation and Content of Premarket Submissions for Device Software Functions, provide guidance about how software should be developed. Others, such as the now outdated 2018 Cybersecurity Guidance, include requirements the software must fulfill, e.g., a user login system, encryption in transit, et al. The former guidance documents should not be listed as requirements because they don’t define what the software must do. The latter guidance documents might be valid.
Even if we assume this requirement as written, it only includes the latter group of guidance—it’s still too big and vague to verify. It should probably be removed or turned into a user need with several specific requirements tracing back to it from the relevant guidance documents.
The system must not allow unauthorized users to log in.
This example was taken from Annex C of AAMI TIR57:2016, Principles for medical device security—Risk management. It states that this requirement isn’t verifiable, since it requires “proving that the system will never allow a particular event, under any conditions.” It then suggests rewording the requirement as follows: “The system must reject unauthorized users.”
I don’t think this is a good argument. Verification doesn’t always need to prove, without any room for error, that the requirement is fully met. At some point you need to stop testing and ship the device so it can begin benefiting patients. Rewording the requirement doesn’t change this. The two requirements are logically equivalent. Any time one requirement fails, the other also fails.
One might suggest rewording this requirement again: “The system must allow users with valid passwords to log in.” This third version could be verified by attempting to log in with an invalid password and a valid password. However, even in this case you don’t know that every password will always let the user log in. Furthermore, presumably you still don’t want users without valid passwords to login too! Thus, the real problem isn’t that a negative requirement is used, but that cybersecurity requirements are difficult to verify with certainty.
Unnecessary Requirements 🔗
The system must be developed in compliance with FDA 21 CFR 820.30.
This is not a valid requirement because 21 CFR 820.30, or the design controls regulation, applies to the design process that was used to develop the software. Thus, it is not really a requirement of the software itself.
The system must include a patient home page.
This requirement is unnecessary. There are almost certainly other requirements that indicate functionality will be present on the patient home page. For example, “The patient home page must list all tests the patient has completed.” This requirement implies that the patient home page exists.
The header bar must show the respective user’s email address and username when users hover over another user’s profile picture.
It depends on additional context, but this requirement doesn’t seem very important. If there’s no verification test for this feature and somehow it breaks, it's probably fine. Furthermore, it’s the sort of feature that’s unlikely to break. Thus, it can probably be removed.
Not every aspect of the software's functionality can or should be listed as requirements. To do this would require, more or less, that you write an SRS that is as detailed as the source code itself. Thus, requirements should be reserved for things that need to be verified.
The system is not to provide an instruction video.
Although this might be a useful assumption for the design team, it doesn’t state anything that’s required and thus is not a requirement.
Negative requirements like this are useful, so we often write them alongside our requirements and then filter them out before we generate documents for the FDA.
Prescriptive Requirements 🔗
1. On the test history table, each test must be editable by clicking on its [edit] icon.
2. Once a test is edited, there must be options to save the entry and close the window.
These two requirements are likely too specific and might constrain the designs available to engineers implementing the requirements. And if the design changes, the requirements will also need to change. It would likely be better to combine these into a single requirement that says: "The Test History Table must provide a means to edit each test in it." It would also be good to clarify what it means for the results to be editable.
Large Requirements 🔗
The software must display the short intake questionnaire if (a) the patient has completed the long questionnaire twice in the last six months, (b) the patient has donated twice in the last year, and (c) the patient has passed a physical exam in the last year; otherwise display the full intake questionnaire.
Not only would this requirement be easier to read if it was split up, but it also would ensure no side cases were missed during verification. As it is now, the verification will need to test at least four cases:
- If (a) is false, then the full intake questionnaire should be given.
- If (b) is false, then the full intake questionnaire should be given.
- If (c) is false, then the full intake questionnaire should be given.
- If (a), (b), and (c) are true, then the short intake questionnaire is given.
One could argue whether or not the other permutations are necessary—you can’t ever test everything!
If you split up the compound requirement, you’ll ensure that each case is covered during verification (i.e., your traceability analysis will identify any requirements not having verification protocols).
Splitting up the requirement is tricky, since the logic is intertwined. Here’s one way to do it:
- The software must display the short intake questionnaire if all of the short intake questionnaire conditions are met, otherwise it will display the long intake questionnaire.
- The software must check that the patient has completed the long questionnaire twice in the last six months as a condition to taking the short intake questionnaire.
- The software must check that the patient has donated twice in the last year as a condition to taking the short intake questionnaire.
- The software must check that the patient has passed a physical exam in the last year as a condition to taking the short intake questionnaire.
To prevent these granular requirements from being too tightly coupled together we must duplicate some of their content. This might be a reason to place them under a shared node in the requirements tree, thus coupling them together. Depending on how you present your requirements, you might also be able to pull some of the duplication up into the parent node of these shared items. For example:
1 Functional Requirements:
...
1.3 The questionnaire selection algorithm
...
1.3.4 must display the short intake questionnaire, unless the patient meets the following condition (in which case it will display the full intake questionnaire):
1.3.4.1 They have completed the long questionnaire twice in the last six months.
1.3.4.2 They have donated twice in the last six months.
1.3.4.3 They have passed a physical exam in the last year.
Revision History 🔗
| Date | Summary of Change |
|---|---|
| 22 April 2022 | Initial Version |
| 30 June 2023 | Updated to reflect the “Content of Premarket Submissions for Device Software Functions” published on 14 June 2023. |
| 26 June 2025 | Added many more details about user needs, including example. |
