On July 14, 2026, FDA posted a warning letter that should make every software device team re-read their change-control SOP. The agency told Nihon Kohden Digital Health Solutions that a software update it shipped in October 2022, documented internally as a letter to file, required a new 510(k). Not because the company skipped the analysis. Because FDA disagreed with one answer, at one decision point, in the company's own regulatory assessment.
It prompted us to ask a bigger question: where does FDA actually draw the line between "document it and ship" and "file a new 510(k)"? Guidance tells you how to run the analysis. Enforcement tells you where the line is. So we pulled every warning letter FDA has published — 5,712 of them, going back to 2015 — and ran AI classification across the full corpus to find every letter where FDA rejected a manufacturer's own modification decision.

What FDA actually said to Nihon Kohden 🔗
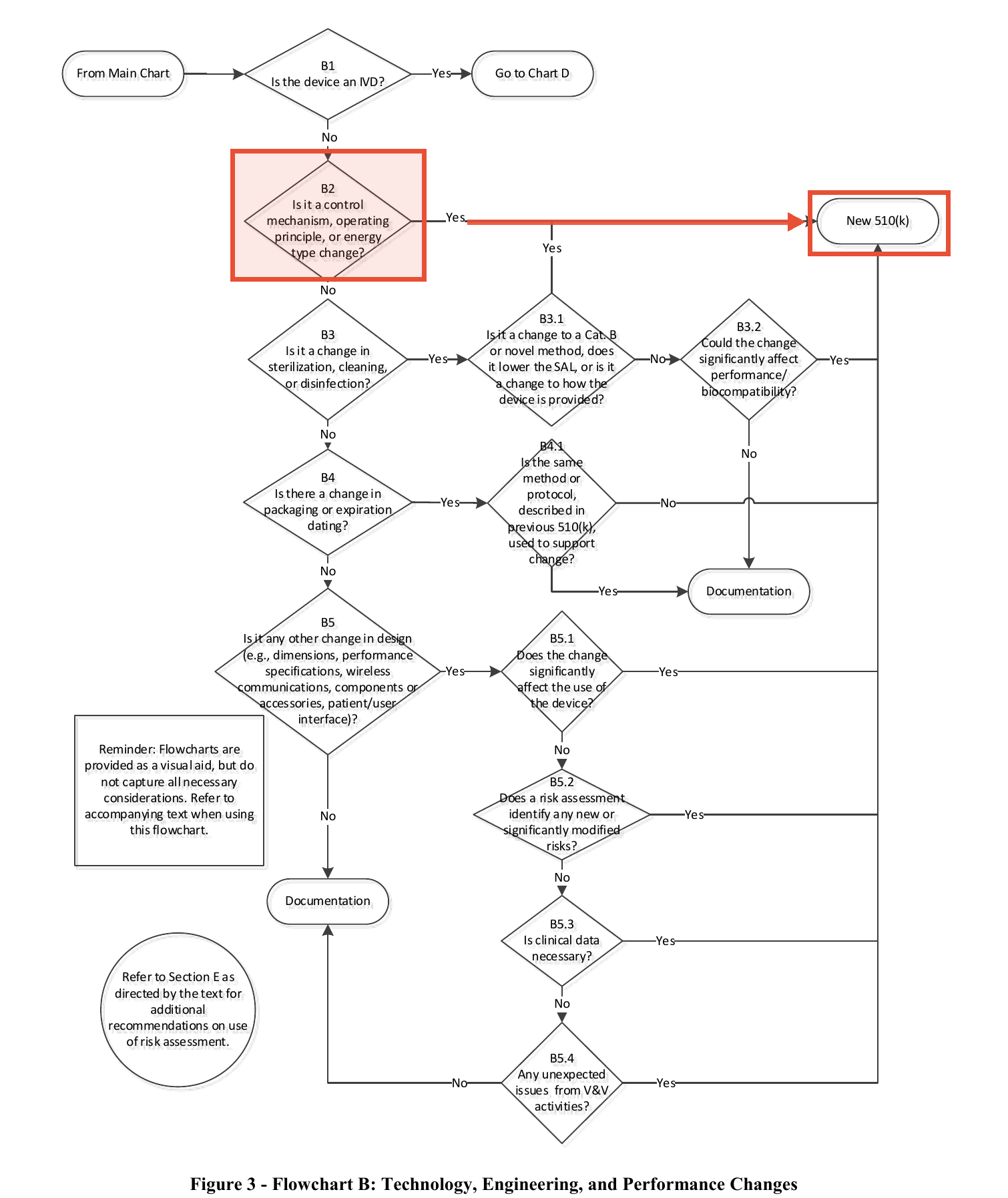
The device is the Next Generation NetKonnect, a patient physiological monitor with arrhythmia detection, cleared as K220989 in July 2022. Three months after clearance, the company added a Silence Alarms function via a software update. Its regulatory assessment walked FDA's own flowchart from the 2017 guidance, Deciding When to Submit a 510(k) for a Change to an Existing Device. At decision point B2 ("Is it a control mechanism, operating principle, or energy type change?") the company answered No. Letter to file. Ship it.
FDA's warning letter quotes that assessment back at them:
"At decision point B2 of the assessment, 'Is it a control mechanism, operating principles, or energy type change?' you selected 'No.' ... However, we disagree with this assessment because adding the Silence Alarms function is a change to a control mechanism of the device and we believe that 'Yes' should have been selected, leading to 'New 510(k).'"
The reasoning: a Silence Alarms function modifies "the manner in which the user controls the device," and a silenced alarm on an arrhythmia monitor can mean a missed life-threatening event. That is a change to a risk control for a significant harm, which is exactly the category the guidance says almost never survives a letter to file. The guidance's own words on B2: "Almost all changes in the control mechanism for a device could significantly affect safety and effectiveness."
There was a second count. The company's labeling described "device-agnostic compatibility": the ability to take input from non-Nihon Kohden devices. K220989 cleared the device for Nihon Kohden inputs only. FDA called that a major change in intended use under 21 CFR 807.81(a)(3)(ii), on top of the control-mechanism change under 807.81(a)(3)(i).
Both counts rest on the same regulation, and it is short. 21 CFR 807.81(a)(3) requires a new premarket notification when a device already in commercial distribution "is about to be significantly changed or modified in design, components, method of manufacture, or intended use," and then defines what qualifies:
(i) A change or modification in the device that could significantly affect the safety or effectiveness of the device, e.g., a significant change or modification in design, material, chemical composition, energy source, or manufacturing process.\ (ii) A major change or modification in the intended use of the device.
Eleven words for the intended-use prong, and no definition of "major." That gap is what the 2017 guidance and its flowchart exist to fill.
Two decisions, both defensible-sounding in a change-control meeting. Both rejected in a two-day inspection in February 2026.
What 5,712 warning letters say about the boundary 🔗
Before drilling into devices, the whole corpus, because a boundary map is only as good as its coverage. Every one of the 5,712 unique letters got the same treatment: full text pulled, product area classified, violation categories extracted. Two-thirds of FDA's warning-letter volume is drugs and tobacco. Devices account for 412 letters (about 7 percent), plus 16 mixed-product letters that also cite device violations, 428 in all. Within those, labeling/misbranding and premarket citations dominate, with quality-system findings right behind. The letter-to-file oversteps live at the intersection of the top two: an unapproved change makes the device both adulterated and misbranded.
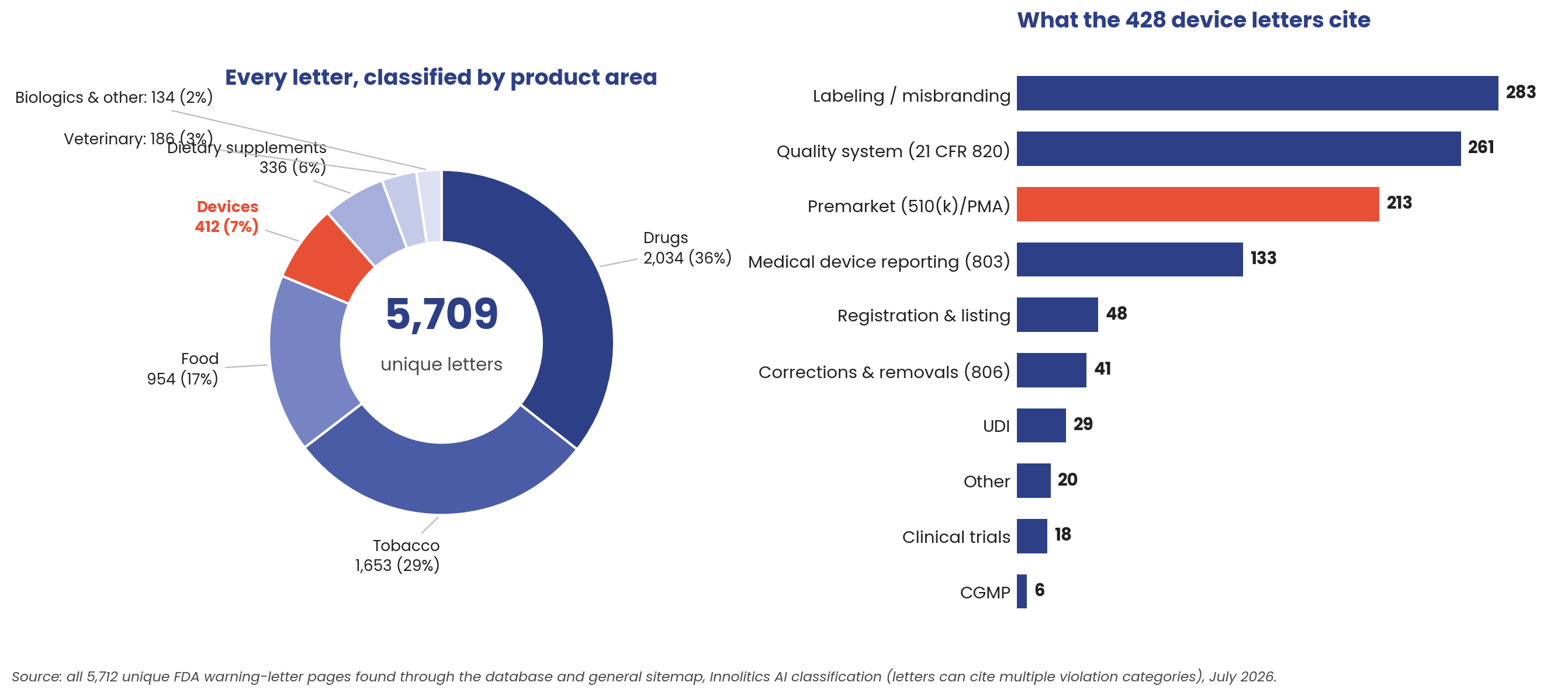
The corpus breaks down like this: 5,712 unique letters, after removing FDA's duplicate postings. 428 concern medical devices. 126 involve a device that is or contains software; only 4 of those explicitly involve AI/ML device software functions. And 86 letters invoke 21 CFR 807.81 against a device that already had a clearance or approval: the modification oversteps. A firm made a change, decided no new submission was needed, and FDA disagreed. 18 of those 86 implicate the device's software: 12 where the change itself was code or firmware, 6 where marketing expanded what the software claims to do.
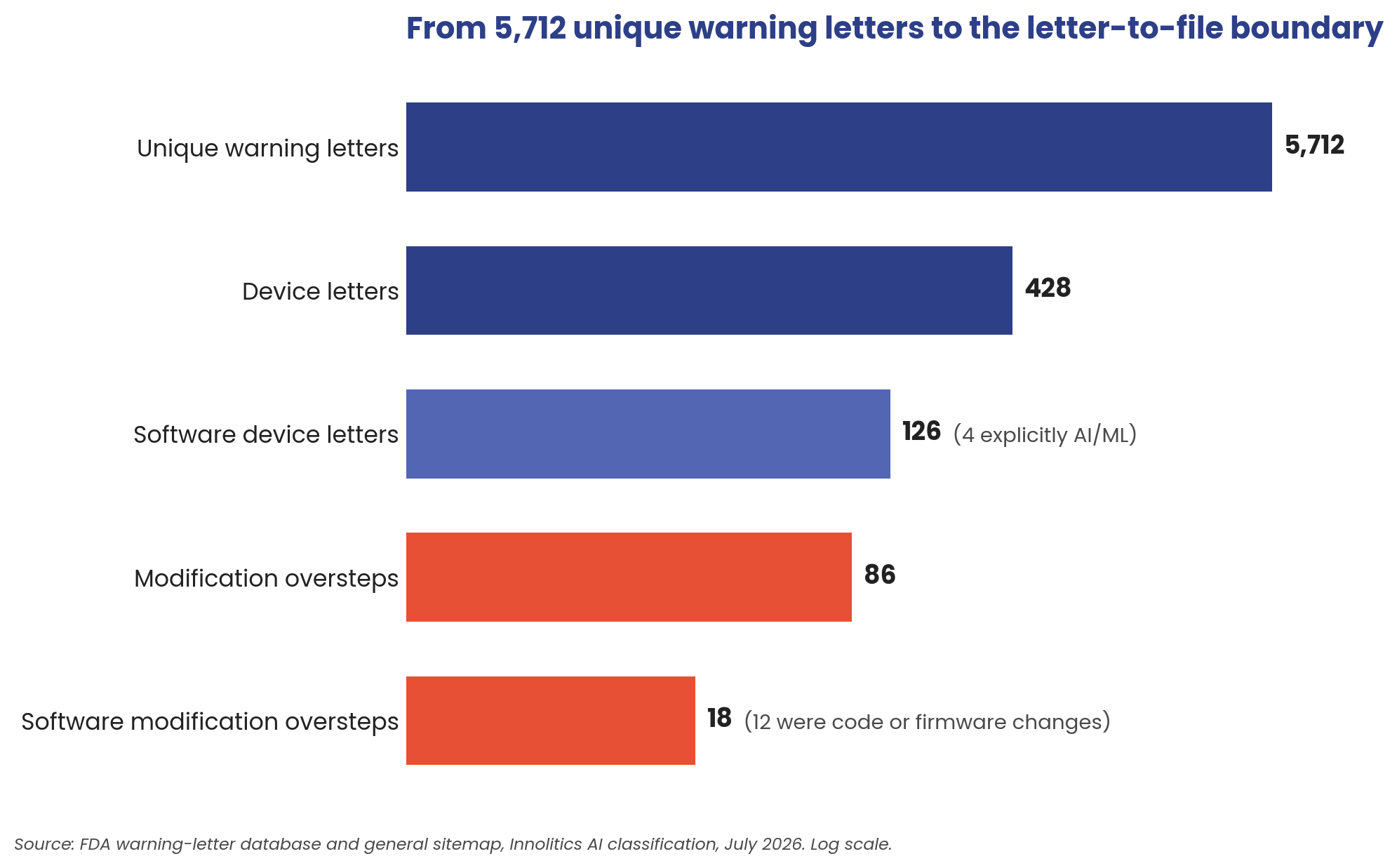
Take the 86 with a grain of salt: warning letters are the tip of the iceberg. Enforcement has three layers. The public layer is warning letters, and they are rare. Beneath it sit the disagreements that end in a 483 observation and get resolved quietly, never becoming public. Beneath that, the changes FDA never sees at all, because no investigator ever opened the file. The warning letters are the egregious cases that have come to our attention, not a census of how often industry gets this call wrong.
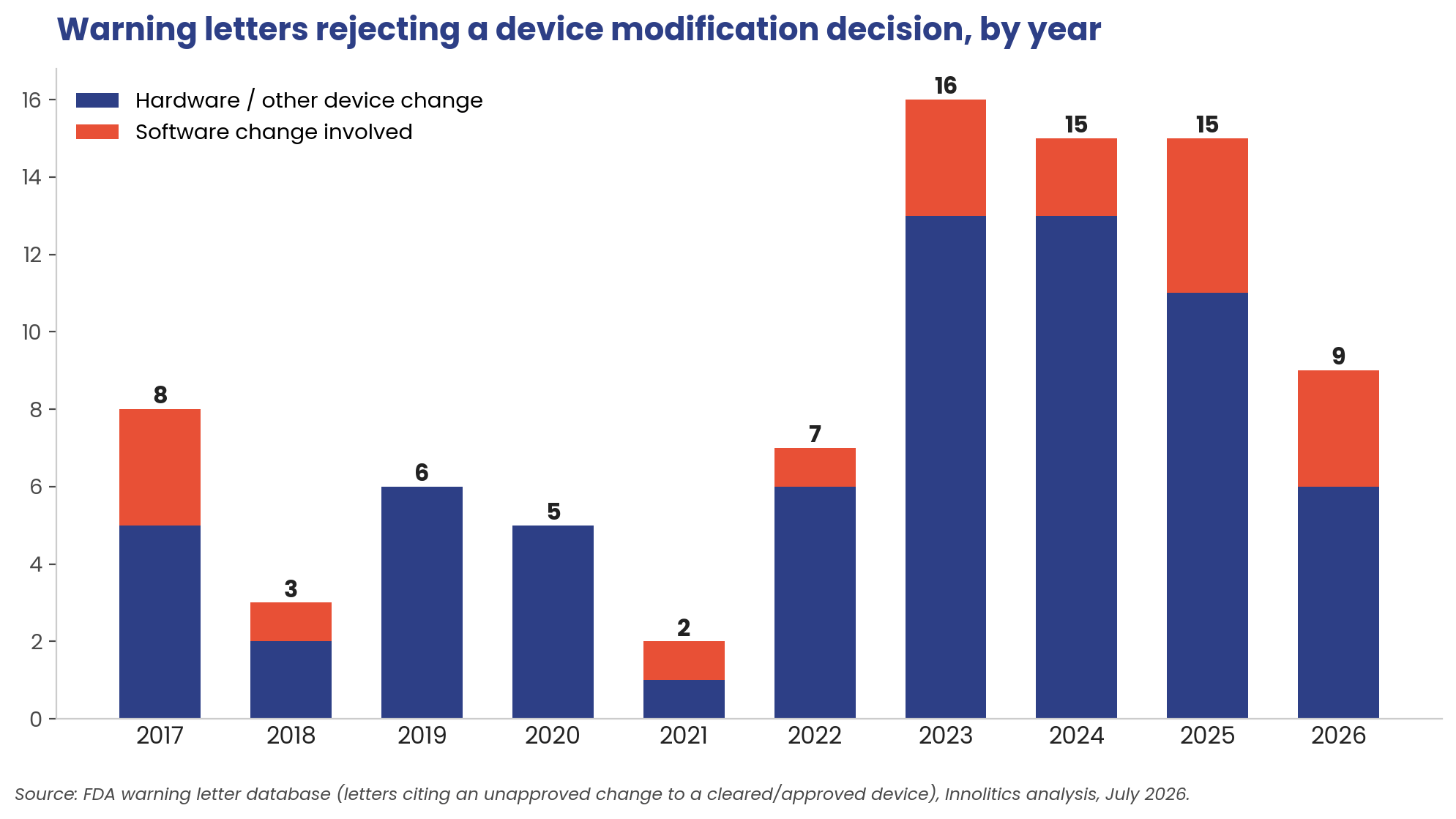
The timing tells its own story. Two overstep letters in 2021. Sixteen in 2023, and roughly fifteen a year since. Nine already in the first half of 2026, on pace for the busiest year in the record. And the software share is climbing: software-implicated changes appear in 7 of the 40 overstep letters from 2021-2024, then 7 of the 24 since January 2025: from under one in five to nearly one in three.
The software calibration table 🔗
Here is every modification overstep in the corpus where the device's software was one subject of the enforcement action: either the change itself was code or firmware, or the company's claims expanded what the software does. What changed, and why FDA said it needed a new submission:
| Year | Company | What changed | FDA's objection | Software the primary finding? | Software-only device (SaMD)? |
|---|---|---|---|---|---|
| 2026 | Nihon Kohden Digital Health Solutions | Silence Alarms function added to patient monitor; "device-agnostic" input claim | Control-mechanism change (807.81(a)(3)(i)); intended-use expansion (a)(3)(ii) | Yes — sole charge in the letter | Yes — NetKonnect runs on off-the-shelf computers |
| 2026 | BMC Medical | CPAP firmware gained new breathing-event detection features (flow limitation, RERA, periodic breathing) | New detection functionality = new 510(k) | Yes — firmware co-equal with hardware and material changes | No — CPAP hardware with firmware |
| 2026 | ZIIP | Microcurrent facial device "re-engineered with advanced technology"; undocumented firmware revisions and app treatment changes | Re-engineering + expanded intended uses required new 510(k) | No — QSR-driven letter; software changes cited as design-control failures | No — handheld microcurrent hardware |
| 2025 | ICU Medical | Infusion-pump software updates, including fixes responding to adverse-event reports | Changes made to address safety problems still require clearance | Yes — sole charge in the letter | No — infusion pump hardware |
| 2025 | Uscom | Spirometer redesign: multi-patient flow tube, Bluetooth, phone app, home use | Technology and use-environment expansion | Partial — software one of three cited changes, plus QSR findings | No — spirometer hardware with companion app |
| 2025 | Red Oak Instruments | Cleared grip-strength dynamometer marketed for concussion screening | Marketing alone changed the intended use | Yes — claims charge leads; MDR/QSR findings follow | No — powered dynamometer hardware |
| 2025 | Technological Medical Advancements | Laser therapy devices with new pulse technology and expanded treatment claims | Technology + indications beyond clearance | No — premarket charge rests on laser tech and claims; software appears only in QSR findings | No — laser therapy hardware |
| 2024 | Xoran Technologies | Cleared CT systems modified: new x-ray receptor panels, software changes, hardware and dimensional changes | Accumulated design modifications required new 510(k)s | No — QSR-led letter; software one line item among many hardware mods | No — CT system hardware |
| 2024 | Cue Health | COVID test substrate, component, and firmware changes (failure detection, temperature correction) | Changes outside the EUA's authorized scope | Partial — firmware co-equal with reagent and component changes | No — reader and cartridge hardware |
| 2023 | iRhythm (Zio AT) | Algorithm and firmware updates; promotion for high-risk, near-real-time monitoring | Performance changes and intended-use expansion | Yes — lead charge; labeling, CAPA, and MDR findings flow from it | No — wearable ECG patch hardware |
| 2023 | ZYTO Technologies | Galvanic skin response device promoted for diagnosing Alzheimer's, HIV, Parkinson's | Diagnosis claims far beyond cleared measurement function | Yes — software disease claims are the letter's reason; QSR tail | No — hand-cradle hardware with companion software |
| 2023 | Vitang Technology | Swapped cleared treatment-planning software for an uncleared version in an aligner system | Component software substitution | Yes — software swap is the sole premarket charge; QSR findings dominate text | No — physical aligner system; the swapped component is standalone planning software |
| 2022 | RightEye | Eye-tracking system promoted for diagnosing neurological disorders | Intended-use expansion via marketing | Yes — claims charge leads; UDI/QSR are add-ons | No — eye-tracking hardware system |
| 2021 | Nikkiso Medical America | Added an adaptive closed-loop controller to a dialysis machine | Autonomous parameter adjustment = new clearance | Yes — sole charge in the letter | No — dialysis machine hardware |
| 2018 | RadLogics | Cleared image-review software (AlphaPoint) promoted with new machine-learning CADe — a “Virtual Resident” auto-detecting nodules, effusions, pneumothorax | Machine-learning detection was a major intended-use change beyond K120161 | Yes — sole premarket charge | Yes — AlphaPoint is standalone imaging software |
| 2017 | Dynavision International | Sensorimotor training board promoted for stroke rehabilitation and concussion management | New therapeutic and diagnostic indications beyond K911938 | Yes — claims charge leads; purchasing/design-control findings follow | No — reaction-training hardware with software |
| 2017 | SyncThink | Eye-tracking system (EYE-SYNC) cleared to measure visual tracking, promoted for concussion and cognitive assessment | Marketing changed the cleared intended use | Yes — sole charge in the letter | No — eye-tracking hardware with analysis software |
| 2017 | Ark Bio Medical | Blood-plasma defroster: obsolete chip replaced, forcing a new circuit board, re-implemented code, and new alarms | Accumulated hardware/firmware redesign required a new 510(k) | Partial — firmware co-equal with the chip, board, and alarm changes | No — plasma-thawing hardware |
A caution before generalizing from the table: not every letter here is about the software. In twelve of the eighteen, the software modification — or the claims expansion about what the software does — is the primary basis of the warning letter, and in seven of those (Nihon Kohden, ICU Medical, Nikkiso, RadLogics, SyncThink, Vitang) it is effectively the only premarket charge. In two (Uscom, Cue Health) the software change is one of several co-equal uncleared changes, and Ark Bio Medical’s firmware sits alongside the chip and board redesign that drove its letter. And in three (ZIIP, Xoran, Technological Medical Advancements) the letter is really a quality-system action that swept software changes in as design-control findings. And only two of the eighteen — Nihon Kohden’s NetKonnect and RadLogics’ AlphaPoint — are software-only devices (SaMD); every other letter concerns software living inside or alongside a hardware device.
1. Changes to how the device intervenes: alarms, controls, autonomy. Nihon Kohden's Silence Alarms. Nikkiso's closed-loop controller. ICU Medical's pump software. Anything that changes how the device acts on or alerts about the patient is a control-mechanism or risk-control change, and FDA treats it as presumptively significant. The ICU Medical letter deserves special attention: some of the software changes were *corrections* responding to adverse events, and FDA still required clearance. Fixing a safety problem does not exempt the fix.
2. New detection or measurement capability. BMC Medical's firmware gained three new breathing-event detections. iRhythm tuned its arrhythmia algorithm and moved toward near-real-time monitoring claims. When the device starts detecting something it did not detect before, or detecting the same thing differently, the cleared performance evidence no longer describes the marketed device.
3. Intended-use expansion, usually via marketing. The most common cluster, and the most seductive, because no code changes at all. Red Oak's dynamometer became a concussion screener on the company website. RightEye's eye tracker became a Parkinson's diagnostic. Nihon Kohden's monitor became "device-agnostic." Uscom's clinic spirometer moved into the home. Your indications are what you promote, not what your 510(k) says, and FDA reads websites.
The iRhythm precedent: what remediation actually costs 🔗
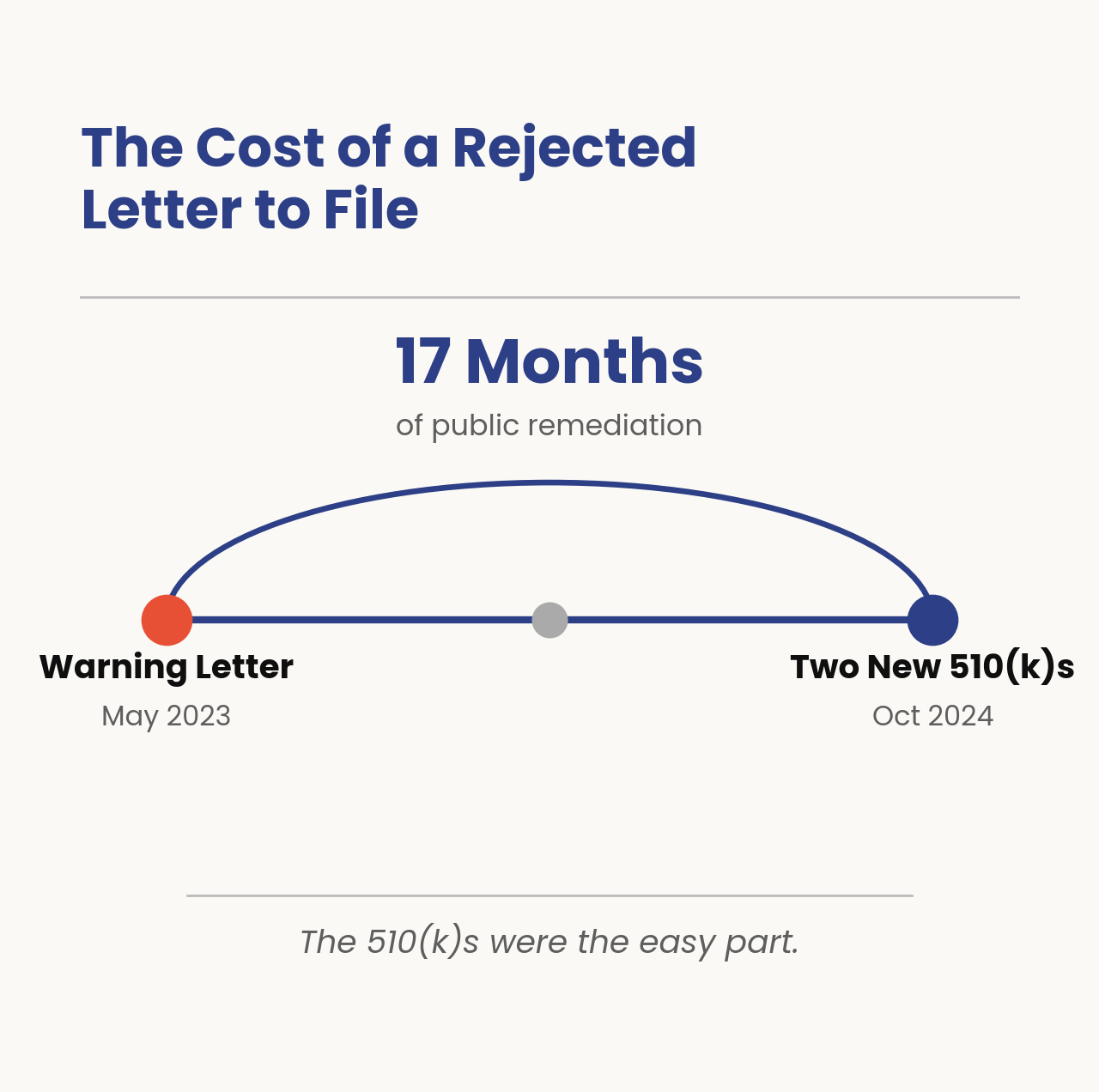
The closest recent precedent shows the price of getting this wrong. iRhythm received a warning letter in May 2023 that, among other findings, cited changes to the Zio AT system (including algorithm adjustments to improve arrhythmia detection performance) that had been documented through letters to file rather than new 510(k)s.
The path back: iRhythm negotiated with FDA, agreed to submit two new 510(k)s covering the accumulated design changes, and received the clearances in October 2024, roughly seventeen months after the letter. Seventeen months of remediation, public disclosure in SEC filings, and a stock price that moved on each announcement, the warning letter was more expensive than just the labor needed to fix it.
Where the boundary actually lies 🔗
Put the guidance and the enforcement record side by side and the letter-to-file envelope is narrower than most engineering roadmaps assume, but wider than regulatory pessimists fear. Eighty-six rejections across eleven years, against tens of thousands of letter-to-file decisions industry makes annually, means the documented no-new-510(k) pathway works. It fails in specific, predictable places.
Stay inside the envelope when the change restores cleared behavior, refactors without touching clinical function, or improves reliability with the same inputs, outputs, thresholds, and claims, with regression evidence to prove it. FDA has never, in this corpus, pursued a company for a well-documented change that preserved the cleared performance envelope and intended use.
You are outside the envelope when any of these is true:
- The change touches a risk control (especially alarms) for a “serious” harm. Alarms, alerts, lockouts, failure detection, fallback behavior. This is B2 territory, and the guidance's own language ("almost all changes in the control mechanism... will usually require submission of a new 510(k)") is the standard FDA applies. Nihon Kohden is the proof.
- The device gains or changes autonomy. Nikkiso's closed-loop controller is the extreme case, but ICU Medical's pump software sits on the same axis. For AI teams: moving from advisory output to anything that acts (auto-triage, auto-titration, auto-silence) is a new device in FDA's eyes.
- Detection or measurement behavior changes. New findings, new event types, retuned operating points. The evidence that cleared the device no longer describes it.
- The claim grows: in code, in labeling, or on the website. Intended-use expansion is the largest cluster in the data, and it is triggered by marketing copy as readily as by software releases. "Device-agnostic," "now for home use," "screens for concussion": each of those phrases, in this corpus, cost a company a warning letter.
And a note for AI teams specifically: only four letters in the entire corpus involve AI/ML explicitly. Three are never-cleared products that marketed diagnosis outright. The fourth, RadLogics in 2018, breaks the easy story: it was a modification overstep on a cleared device — a firm that bolted machine-learning CADe onto image-review software cleared only to display and measure, and marketed it as a “Virtual Resident.” The precedent for an AI modification overstep already exists; it is just eight years old and pre-dates most of the field. What still has not happened is a warning letter for retraining a cleared model — changed data, changed weights — under a letter to file. While the iRhythm letter cites a Zio AT algorithm “adjusted based on available data” — every readable clue (the “events that meet criteria” trigger language, FDA’s citation of the performance-specification branch of the modifications guidance, and the on-patch firmware context) says threshold tuning rather than just retraining, but the (b)(4) redactions leave a sliver of doubt.
But don’t take it to mean this is a “get out of jail free” card. It could very well be just because the AI device fleet is young, inspections lag by years, and the enforcement pattern above (risk controls, autonomy, detection behavior, claims) maps directly onto what a model update changes.
One more pattern worth naming: the clock does not protect you. Nihon Kohden's change ran three and a half years before the inspection.
Takeaways 🔗
Read your own change assessments the way an investigator will. The Nihon Kohden letter is a template: FDA pulled the company's regulatory assessment, found the decision point, and litigated the single checkbox.
Some concrete calibration from the enforcement record:
- Anything that changes how a user controls alarms, alerts, or notifications is a control-mechanism change. Silence, snooze, re-prioritize, auto-dismiss: on a monitoring device, these are risk controls for the harm the device exists to catch. Assume B2 is Yes.
- Compatibility expansion is intended-use expansion. Depending on the technological characteristic, "Works with more inputs" reads could be crossing the line. If the cleared device supported a defined input set, opening it up (device-agnostic, vendor-neutral, third-party) is a special or traditional 510(k) trigger.
- Algorithm performance tuning is not maintenance. iRhythm's letter treated detection-performance adjustments as reportable changes requiring new evidence. If the operating point moves, the burden of proof moves with it.
- The absence of complaints is not evidence. Nihon Kohden's change ran in the field for over three years before an investigator read the file. Time in market does not ratify the decision.
- A PCCP is the escape hatch: before the fact, not after. Alarm-behavior changes and input-set expansions are exactly the kinds of modifications a well-drafted PCCP could have authorized in advance. That option exists only if you negotiate it into a clearance before you need it.
The companies that are too conservative run out of cash before they get to market. The companies on the opposite end of the spectrum fail just as badly with enforcement action or reputational damage due to warning letters or patient safety issues. The science and art of all this is figuring out the right balance that works for you and your company.
Innolitics helps AI and software medical-device teams make exactly these calls (change assessments, PCCP strategy, and 510(k) submissions) before a release decision becomes a compliance problem. If your team is sitting on a stack of letters to file and wondering how they would survive an inspection, that review is a lot easier now than with an FDA inspector with a gun at the hip and a badge at the door.