











Walk into the next board meeting with a guaranteed FDA timeline.
Four weeks to a plan and budget. A guaranteed date to a submission-ready 510(k). The same team that builds the device commits to the date. You commit it to the board.
Recent case study
We Did It All: R&D, Clinical Study, and 510(k) Cleared in 12 Months
Built and cleared an AI cardiomegaly detector end-to-end: algorithm R&D to 510(k) clearance in under 12 months.

We went from idea to FDA clearance in 18 months and first Install for AI Tumor Tracking Software
Prototype → 510(k) → QMS → FDA audit passed. Multi-year partnership with ongoing development.

Competitor: 5 years, $500k, no results. Us: 18 months, FDA cleared, IPO filed.
Full-cycle development of GyriCalc, a first-of-its-kind pediatric neuroimaging diagnostic. FDA cleared (K250686) July 2025. Filed for IPO in June 2025.

Recent end-to-end engagements
Our Clients Include
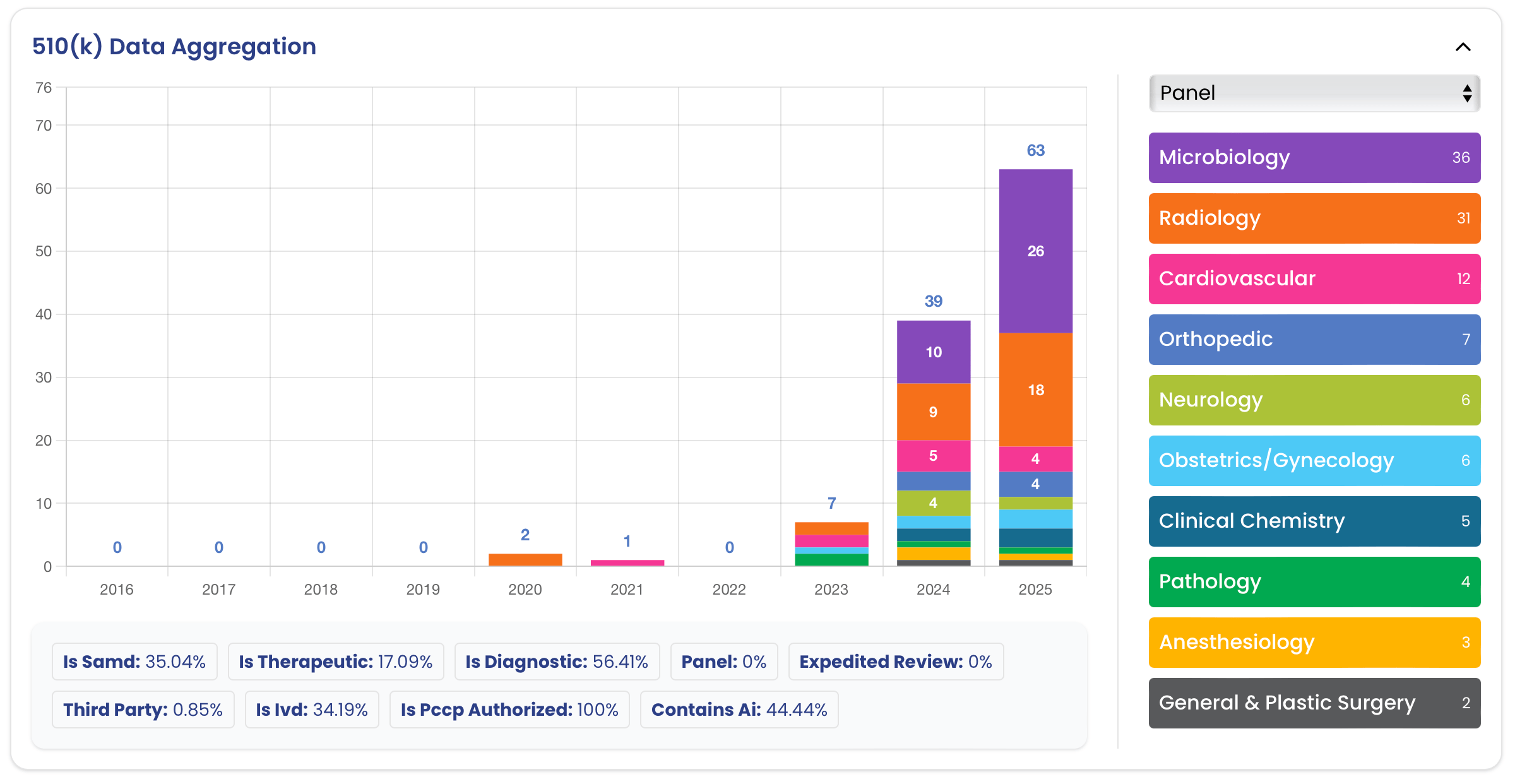
FDA Clearances Include
-
AI
-
AI

Salix Coronary Plaque
-

Lightning Viewer
-
AI
GyriCalc
-
AI

Galileo CDS GBrain MRI
-
AI

Neosoma Brain Mets
-
AI

BioticsAI
-

Zeto New Wave System
-
RadUnity
-
AI

Galileo CDS
-
AI

Prenuvo
-
AI

TOMI Scope
-

Rology Teleradiology
-
AI

DiA Imaging LVivo Software Application PLAX module
-
AI

Echo IQ
-
AI
Salix Central
-
AI

Automatic Anatomy Recognition Software
-
AI

Limbus Contour
-

MD.ai
-

Mobius3D
-
AI

Corticometrics
-

FlexView
-
AI

Specific Dx
-
AI

Envisionit Deep AI
-
AI

Cube Click
-
AI
AI Metrics
-
AI
CT Cardiomegaly
-
AI

SimBioSys
Is this for you?
You need a one stop shop. Not another email.
Founder or CEO without internal SaMD depth
You have data, a clinical intent, and urgency.
We own concept to clearance. You stay on clinical and commercial. Your board sees a real timeline, not a slide deck.
Funded AI/ML medical device company
You need to compress development, validation, and submission.
We close the seams between engineering, AI/ML validation, cybersecurity, and regulatory so the timeline shortens without creating regulatory debt downstream.
Stalled or burned program
You have a codebase, a failed submission, a hold letter, or a vendor mess.
We triage what is salvageable, take engineering and regulatory to the finish line, and do not restart what does not need restarting.
Proof, not promises
Recent FDA wins.
Every case below had Innolitics on both the engineering side and the regulatory side. No vendor handoffs. One team, one outcome.
You just saw six clearances.
Yours could be the seventh.
Send the device, the indication, and your target FDA center. We will tell you whether it looks like the ones above and what your version of that timeline would be.
The core differentiation
We remove the seam between engineering and regulatory.
The failure point is the seam. Programs do not fail because one workstream is impossible. They fail because engineering evidence, AI/ML validation, regulatory strategy, cybersecurity, and quality records get built by different teams with different incentives.
Innolitics puts software engineers, AI/ML scientists, MDs, PhDs, regulatory leads, and quality specialists on the same team, on the same Slack, on the same deadline. You give us the clinical intent, the data, and the target FDA center. We give you a cleared device. Submission package, V&V evidence, cybersecurity dossier, labeling, the whole design history file. 45+ submissions and counting.
And because we own the seam, we can stand behind the outcome. The team delivering the work is the same team committing to the date.
For devices that qualify after a paid four-week planning phase, we put a clearance timeline and success guarantee in writing. Few firms in this space will sign that.
What 'end-to-end' actually includes
One accountable team. Eight workstreams. One clearance path.
Wrangling multiple vendors is like herding cats. But unlike cats, your company does not have nine lives. You have one shot with FDA. Make it count.

Product & Software Engineering
Working SaMD that can support FDA evidence, not just a prototype. Frontend, backend, DICOM, FHIR, workstation integrations. IEC 62304 Class C where it has to be.
AI/ML Model Development
Training, testing, generalization, and clinical performance evidence. PyTorch, TensorFlow, MONAI, MRMC reader studies.
Quality Management System
Design controls and records that hold up under FDA audit. Stand up a new ISO 13485 QMS, or plug into yours.
Regulatory Strategy & Submissions
510(k), De Novo, Pre-Sub, PMA supplements, BDD. We write, we submit, we respond to FDA.
Verification & Validation
Requirements-to-test traceability and defensible evidence. Unit, integration, system, and clinical performance.
Cybersecurity (FDA 2023)
SBOM, threat modeling, SPDF, vulnerability handling. The new guidance, done right the first time.
Risk Management (ISO 14971)
Hazard analysis, FMEA, benefit-risk reasoning. Tied into design, V&V, and post-market from day one.
Post-market & PCCP
Predetermined Change Control Plan, post-market surveillance, SaMD model monitoring.
Not in scope: clinical trials, wet lab operations, sales, or work the sponsor intentionally keeps in-house. Everything else between your data and a clearance letter is ours.
How an end-to-end engagement works
Five steps. One accountable team across all of them.
We do not hand off between phases. The same team scopes, builds, submits, and supports the device through clearance and beyond.

-
1
Fit and pathway assessment
Confirm device type, clinical intent, data readiness, target FDA center, likely pathway, and major risks. We tell you whether we are the right partner.
-
2
Program architecture
Define product scope, regulatory strategy, QMS structure, validation plan, cybersecurity plan, and a real timeline you can defend to your board.
-
3
Build and document
Develop software, AI/ML model, evidence, design history file, submission artifacts, and V&V records together, not sequentially.
-
4
Submit and respond
Submit the package, handle FDA questions, refine evidence, and drive toward clearance. We write the response. You sign it.
-
5
Transition or continue
Support post-market obligations, PCCP, maintenance, repeat indications, or hand off to your in-house team. Your call.
Which device category looks like yours?
If it has an algorithm, we have probably already done it.
Six SaMD archetypes below: radiology AI, specialty imaging, cardiac AI, workflow SaMD, radiation oncology, and connected hardware-plus-software. Pick the row that looks most like yours.

Radiology AI
Radiology CADe, CADx, and segmentation
Detection, quantification, or measurement software against imaging. Reader studies, MRMC, AI/ML model validation, PCCP, and 510(k) under the FDA's AI/ML guidance.
Imaging
Specialty imaging: pathology, dental, OCT, ophthalmology
Whole-slide pathology AI, dental panoramic CADe, OCT analyzers, fundus AI. Clinical performance studies, color and modality calibration, DICOM, and clinical workflow integration.
Cardiac AI
Cardiac and physiological-signal AI
ECG, echo, and physiological-signal AI. Foundation models on cardiac signals, Breakthrough Device strategy, 510(k) and De Novo pathways, and Pre-Sub work that lands.
Workflow
Workflow and DICOM-based SaMD
Image routing, viewers, structured reports, teleradiology, and integration SaMD. Orthanc, FHIR, HL7, cybersecurity, and 510(k) for software that orchestrates clinical work without making a diagnostic call.
RadOnc
Radiation oncology and treatment planning
RT planning, dose verification, CBCT QA, brain-mets segmentation, and oncology decision support. CDRH submissions for software that touches dose, with multi-year platform partnerships.
Connected
Connected hardware and software medical devices
Device firmware, embedded software, cloud and mobile companions, and the SaMD layer for hardware-first products. IEC 62304 Class C, cybersecurity (FDA 2023), and 510(k) submissions where the software is the regulated article.
Before we put a date in writing
A four-week planning phase. A real answer.
A paid four-week planning phase to pin down your data, predicates, and pathway. The deliverable is a scoped program with a clearance timeline we will guarantee. If the device does not qualify, we tell you that too.
What Our Clients Say
The lines clients repeat on reference calls.
After spending $500K over five years on consultants who knew how to sell but couldn't execute, Innolitics took us from concept to FDA clearance in under 18 months for a first-of-its-kind MRI neuroimaging device. When FDA questioned our original approach, they adapted without losing momentum. We submitted on schedule and cleared in 4.5 months. I've seen many engineers at J&J and Ethicon — Innolitics is many standard deviations ahead of the curve.

Andrew Stewart
CEO Neuro Spectrum Insights
Before you book the call
First-call questions we hear most.
What does 'end-to-end' actually mean? What is in scope?
Everything between your clinical intent and a clearance letter. Product and software engineering, AI/ML model development, quality system, regulatory strategy and submission, V&V, cybersecurity, human factors, risk management, and post-market. What is not in scope: your clinical trials, your wet lab, your sales force, and the things you would rather keep in-house. We map the line on the first call.
What do we need to have before we engage?
For the first call: a device concept, a sense of clinical intent, and any data or code you have already collected. That is enough. For the engagement itself, what ‘ready’ looks like depends on the device. For a 510(k) AI SaMD, you typically need a defensible training set, a held-out test set that reflects the intended use, and ground truth from qualified readers. If you are not there yet, we scope the data collection and curation work as the first phase. The first call exists to figure out where you actually are, which is usually different from where you think you are.
We already have the model trained. Do we still need you?
Yes, and we can come in regulatory-only. About a third of our engagements arrive that way: a working model, partial documentation, and an FDA pathway to figure out. We own regulatory strategy, V&V evidence, QMS gaps, cybersecurity dossier, the 510(k), and the FDA response from there, without rewriting code that already works. The clearance date guarantee still applies after the planning phase. (And if you want us to build the model from scratch, we do that too. AI Metrics and SmileDx both came in that way.)
Who controls the FDA relationship and the IP?
You do, on both. Your regulatory lead signs the submission and remains the official sponsor contact. We draft, prepare you for the call, sit in on the meeting, and write the FDA responses. You own the source code (your repos), the model weights (your infrastructure), the design history file (your QMS), and the submission itself. We are the team that builds it. You are the sponsor.
How long, how much, and do you guarantee the date?
Timeline: BodyCheck went idea-to-510(k) cleared in 12 months including the clinical study. AI Metrics was 18 months. GyriCalc was a similar magnitude. Regulatory-only engagements move faster: MedWeb was 7 weeks from kickoff to submission and 6 months to clearance because the software was already built and we owned the regulatory work end to end. If you are well-funded with training data ready, we can compress an end-to-end build to about 9 months.
Price: fixed-fee milestone pricing where we can scope cleanly; time-and-materials where scope is genuinely unknown.
Guarantee: for devices that pass the paid four-week planning phase, we put a clearance timeline and success guarantee in writing. The planning phase exists for a reason. We will not commit to a date until we have looked at the data, predicates, and pathway closely enough to stand behind the number.
How does the guarantee actually work?
After the paid four-week planning phase — once we have looked at your data, predicates, and pathway — we put two commitments in writing. The Timeline Guarantee: your software and submission documentation will be submission-ready by the committed date, or you receive a partial refund. The Clearance Guarantee: if FDA issues a Not Substantially Equivalent decision on work that was within our scope, you receive a partial refund. The clock pauses for things outside our control — FDA delays, unpaid invoices, or when we are waiting on your team. We only sign the guarantee for devices that clear the planning phase, because we will not put a number in writing that we have not stress-tested.
Can you help us fix a failed submission or an FDA hold letter?
Yes. SimBioSys came to us with a hold letter; we found the response that landed the clearance without a new clinical study. Neosomas came to us with a monolithic first submission and wanted a better architecture; we designed one. Rescue engagements are Option B in the engagement models above.
When is Innolitics not the right fit?
When you want a binder of advice rather than a built product. When you need clinical trial operations or wet lab work, which we do not do. When the device is genuinely outside SaMD or SiMD, for example a hardware-first implant or a pure drug-device combination where the drug is the regulated article. And when the timeline is shorter than physics allows: we will tell you on the first call rather than take the engagement.
30 minutes. No deck. No follow-up emails.
Book the fit call. Walk away with an answer.
Pathway, timeline, missing workstreams, and an honest go or no-go on whether Innolitics would take the engagement. If we are the wrong fit, you save months.
What CEOs read before booking the call
We know our stuff.
Innolitics editorial grouped by the questions executives ask before they engage. Pathway selection, FDA pre-submissions, AI/ML validation and acceptance criteria, cost, and the post-clearance platform play.

Should we use 510(k) or De Novo?
De Novo Requests for Diagnostic Devices: FAQs and Examples
A concise guide to the FDA’s De Novo pathway for novel, moderate-risk medical devices that lack a suitable predicate. It includes real-world exampl...
 J. David Giese
J. David Giese

How do we engage FDA early?
FDA Pre-Subs: Best Practices, FAQs, and Examples
A Pre-Sub is a mechanism for requesting formal written feedback from the FDA, and (optionally) a one-hour meeting. Pre-subs are a useful means to m...
J. David Giese
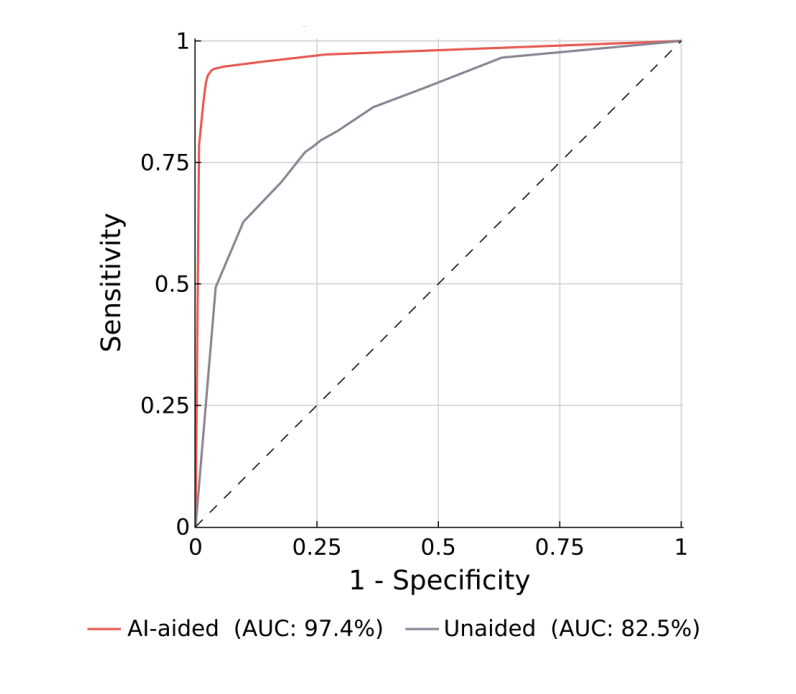
What validation will FDA expect?
Reference Guide for Testing Expectations for AI/ML-Enabled SaMD
A “quick” reference guide grouping the example 510(k) clearances by their product code/regulation, summarizing required testing and the rationale b...
 Meri Martinez
Meri Martinez
What does FDA clearance actually cost?
How much will an FDA clearance cost?
For founders, CEOs, and CFOs at AI/ML medical device companies trying to decide how much to spend on a 510(k). A meta-analysis of 35 post-IPO AI/ML...
 Yujan Shrestha
Yujan Shrestha

How do we set AI/ML acceptance criteria?
Definitive Guide to AI/ML SaMD Acceptance Criteria
Setting acceptance criteria incorrectly (too high or too low) can delay your FDA submission by weeks or trigger rejection. This guide analyzed 784 ...
Yujan Shrestha
How do we keep updating the model after clearance?
PCCPs: Best Practices, FAQs, and Examples
With predetermined change control plans (PCCPs), FDA has given manufacturers a great new regulatory tool. In this article, we discuss PCCP best pra...
J. David Giese
Let's Talk
Every great partnership starts with a conversation. Fill out the form below for a discovery call, and an Innolitics team member will contact you soon.
I learned more from an hour-long Innolitics sales call than an entire paid engagement from other consultants.

Richard Clark
Executive in Residence at Washington University