![]()
In early 2025, the software engineering world was introduced to a new term that quickly moved from a viral social media post to the Merriam-Webster dictionary: vibe coding. Coined by AI researcher Andrej Karpathy, the term describes a paradigm where the engineer specifies intent in natural language and accepts the AI's generated code with light or no manual editing [1]. The human acts as the director; the Large Language Model (LLM) acts as the author. Tools like Cursor, Claude Code, ChatGPT Codex, Lovable, Replit, Antigravity, GitHub Copilot, and Windsurf have put this paradigm in reach of anyone who can describe what they want, from solo clinicians prototyping algorithms to enterprise teams shipping production software.
But the paradigm has already accelerated beyond vibe coding. We are now entering the era of Agentic Code, where autonomous AI agents (Devin, Aider, the agentic modes inside Claude Code and Cursor, Bolt, v0, and increasingly capable IDE-native agents like Antigravity) write, test, and deploy software iteratively without human prompting. The byproduct is Dark Code: any code that no human eye has actually read.
For developers building consumer web apps, this autonomous generation is a revelation. But for engineers and regulatory affairs professionals in the medical device industry, the concept of "Dark Code" triggers immediate alarm bells. Medical device software must comply with rigorous standards: IEC 62304 for software life-cycle processes, ISO 14971 for risk management, and ISO 13485 for quality management systems [2] [3].
The prevailing sentiment among many regulatory consultants is that a vibe-coded codebase is inherently incompatible with FDA clearance. They argue that AI-generated code is too opaque, too prone to unverified dependencies, and too detached from traditional requirements traceability to ever pass regulatory muster.
I disagree.
In this article, I'll explore the data behind AI/ML Software as a Medical Device (SaMD) clearances, examine historical precedents that prove the FDA's adaptability, and outline the exact steps required to bring a vibe-coded prototype to full regulatory specification.
The SaMD Landscape in Numbers 🔗
Before discussing how to clear a vibe-coded or autonomously generated device, understand the current state of Software as a Medical Device (SaMD) clearances. Using the Innolitics FDA Browser, I analyzed every SaMD clearance from 2016 through April 2026. The data reveals a regulatory environment that is not only highly active but also increasingly comfortable with complex software architectures.
The Exponential Rise of SaMD Clearances 🔗
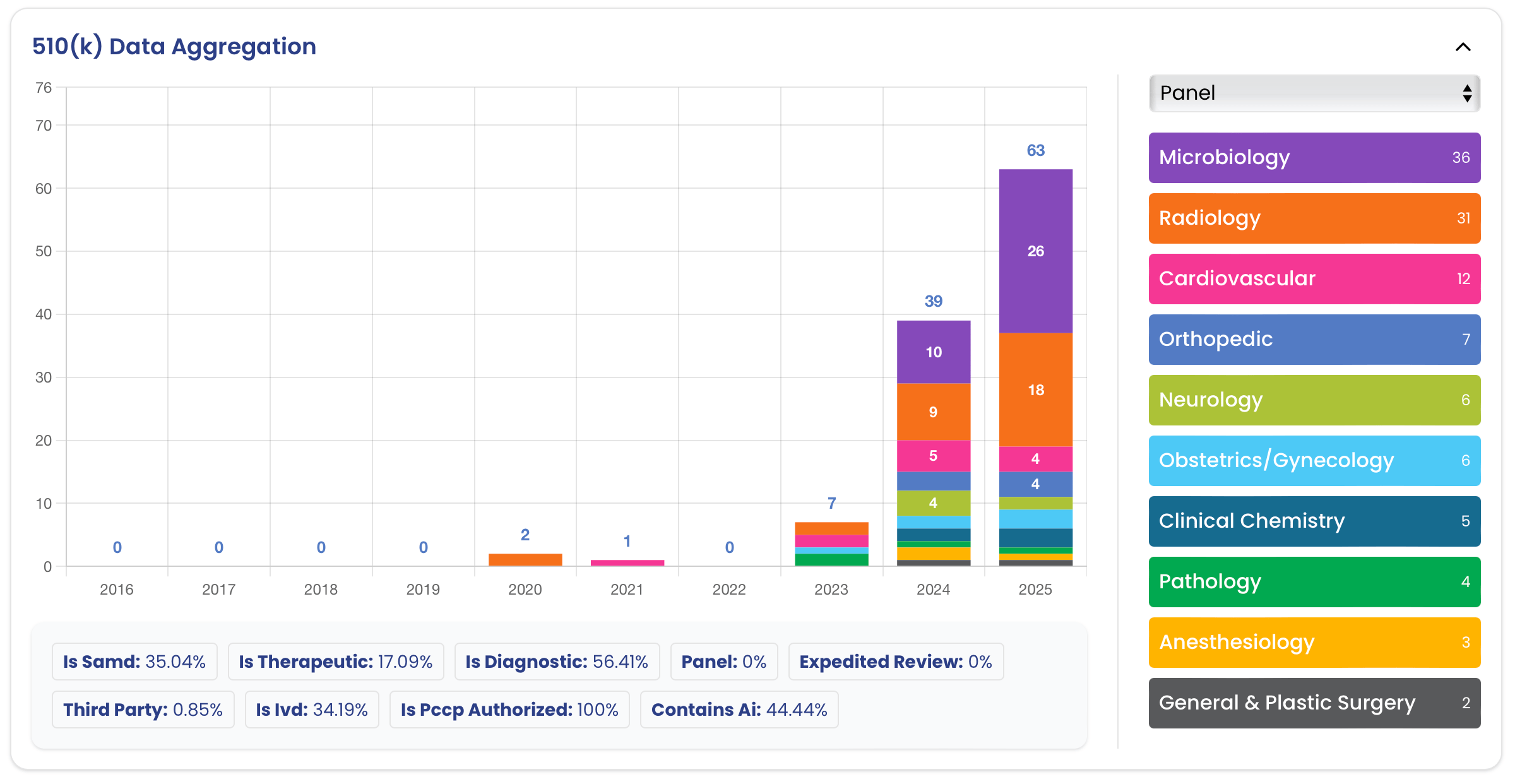
The FDA has cleared 2,589 SaMD devices since 2016. Of those, 963 (37.2%) are AI/ML-enabled. As Figure 1 illustrates, the volume of SaMD clearances is climbing rapidly, with 2025 setting a new record of 390 clearances, and AI/ML now making up 59% of that annual total.
This growth is not limited to a single medical specialty. While Radiology remains the gravitational center of SaMD, accounting for roughly 63.6% of all clearances, other panels such as Cardiovascular, Clinical Chemistry, and Neurology are steadily gaining ground (Figure 2).
Historical Precedent: The FDA Has Seen This Before 🔗
The skepticism surrounding AI-generated code echoes the skepticism that accompanied previous shifts in software engineering. Time and again, the industry has assumed that a new development paradigm would be rejected by the FDA, only to find that the agency's existing frameworks were flexible enough to absorb the change.
As Figure 4 shows, the FDA has already cleared hundreds of devices utilizing paradigms that were once considered "too modern" or "too different" for medical use at the time. Let's examine a few of these precedents.
Command-Line Interface as a Medical Device (CLaMD) 🔗
Historically, medical device software was expected to have a Graphical User Interface (GUI). The GUI was where the clinician interacted with the device, and therefore, it was the focus of extensive human factors engineering and UI hazard analysis.
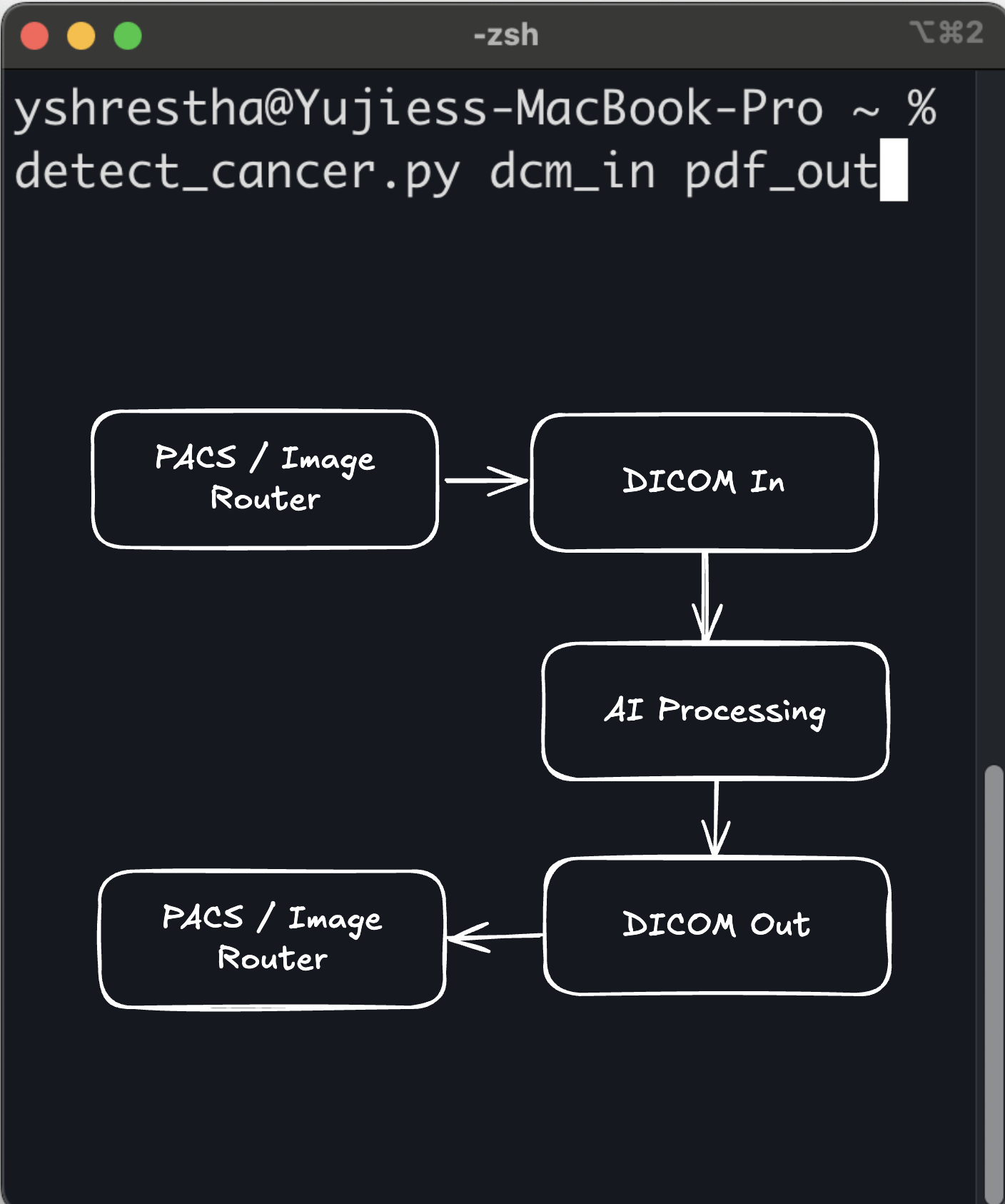
When developers began submitting "headless" algorithms that operated entirely via a Command-Line Interface (CLI), taking an input file, processing it, and returning an output file without any user interaction, many assumed the FDA would balk. How could you clear a device that the clinician never actually sees?
As I detailed in my previous article on Command-Line as a Medical Device, the FDA accepted the CLI paradigm readily. The regulatory logic is elegant: a CLI shifts the risk-control burden for display, navigation, and confirmation steps to another device (such as a cleared PACS or EHR system) that is already authorized for those tasks.
- Thirona LungQ 4 (K250766, Oct 2025): "LungQ is a docker image with a standalone command-line software which must be run from a command-line interpreter and does not have a GUI."
- Nurea PRAEVAorta®2 (K243859, Aug 2025): "Input of patient data: Command line interface (API)."
- Brain Electrophysiology NEAT 001 (K250058, Apr 2025): "Sleep stages are scored by the containerized neat-cli software on the FLOW server."
The CLI precedent proves that the FDA does not require software to look or behave like a traditional desktop application. It only requires that the software's inputs, outputs, and risks are strictly defined and verified.
Software Development Kit (SDK) as a Medical Device 🔗
An even more abstract paradigm is the Software Development Kit (SDK) or software library. An SDK is not a standalone application; it is a collection of code intended to be integrated into another developer's application.
- Measure Labs / Preemptive AI Clinical SDK (K250233, Feb 2026): "SaMD SDK for integration into third-party mobile apps; operates on Android/iOS."
- Nobel Biocare DTX Studio Assist (K252086, Nov 2025): An SDK with no UI, bundled with host dental imaging software.
- Deepwell DTx ABS (K233580, Aug 2024): An SDK for Android breathing biofeedback.
Agile and Iterative Development 🔗
For two decades, the medical device industry treated waterfall as the only acceptable software lifecycle. Gather all requirements upfront. Freeze the design. Code. Verify. Ship. Anyone proposing two-week sprints was told it would never survive an FDA audit.
That assumption was wrong.
AAMI TIR45 (Guidance on the use of AGILE practices in the development of medical device software) was published in 2012 and is recognized by the FDA in its consensus standards database. It maps Scrum, Kanban, and continuous integration directly onto IEC 62304 lifecycle activities. Each sprint becomes a planned design iteration. Each release passes verification. The QMS captures the trail.
Read the underlying standards and the truth is obvious: IEC 62304 never required waterfall. It required lifecycle activities: planning, requirements, architecture, implementation, verification, validation, risk management, configuration management, problem resolution. It is silent on cadence. Whether those activities span a 24-month waterfall or a 2-week sprint is a methodology choice, not a regulatory one.
Here are some examples where the design methodology is publicly available in the 510(k) summaries. Countless devices likely use iterative development but do not publicly state it.
- PathAI AISight (K243197, Jul 2025): Submission documents Scrum-based iterative development with sprint-aligned design reviews under IEC 62304.
- Aidoc BriefCase (K251884, Sep 2025): Continuous integration pipeline with automated regression testing as the verification spine across releases.
- GE HealthCare Critical Care Suite (K252002, Oct 2025): Iterative release cadence governed by a PCCP, with each release re-verified against the locked intended use.
Agile got cleared because the FDA was never asking for waterfall. It was asking for evidence. Sprints, backlogs, and continuous integration are a different cadence for producing the same artifacts: requirements, design, verification, risk controls, traceability. The methodology shifted; the regulatory deliverables did not.
Containerization and Modern Stacks 🔗
When Docker and containerization revolutionized software deployment, the medical device industry was hesitant. Could a containerized microservice be cleared, or did the FDA require monolithic, bare-metal installations?
- Clouds of Care PreOp v3 (K252565, Feb 2026): Cleared with a "modular, containerized software architecture that replaces the monolithic design of PreOp V1."
- TeraRecon Cardiovascular.Calcification.CT (K250288, Oct 2025): Described as a "containerized application (e.g., Docker)."
Similarly, the shift from legacy languages like LabVIEW to modern stacks like Python has been seamlessly absorbed. The AccurECG Analysis System v2.0 (K252361, Dec 2025) successfully used a LabVIEW predicate to clear a Python rewrite.
The Innolitics Thesis: Wrap, Don't Rewrite 🔗
The core argument against AI-generated code is that an LLM cannot produce code that is inherently compliant with IEC 62304. This is true. An LLM cannot generate a complete, traceable risk file or a verified software architecture document out of thin air.
However, this argument misses the point. No code is inherently compliant.
A human engineer writing Python in a text editor is not producing compliant code either. Compliance is not a property of the code itself; it is a property of the evidence and provenance surrounding the code.
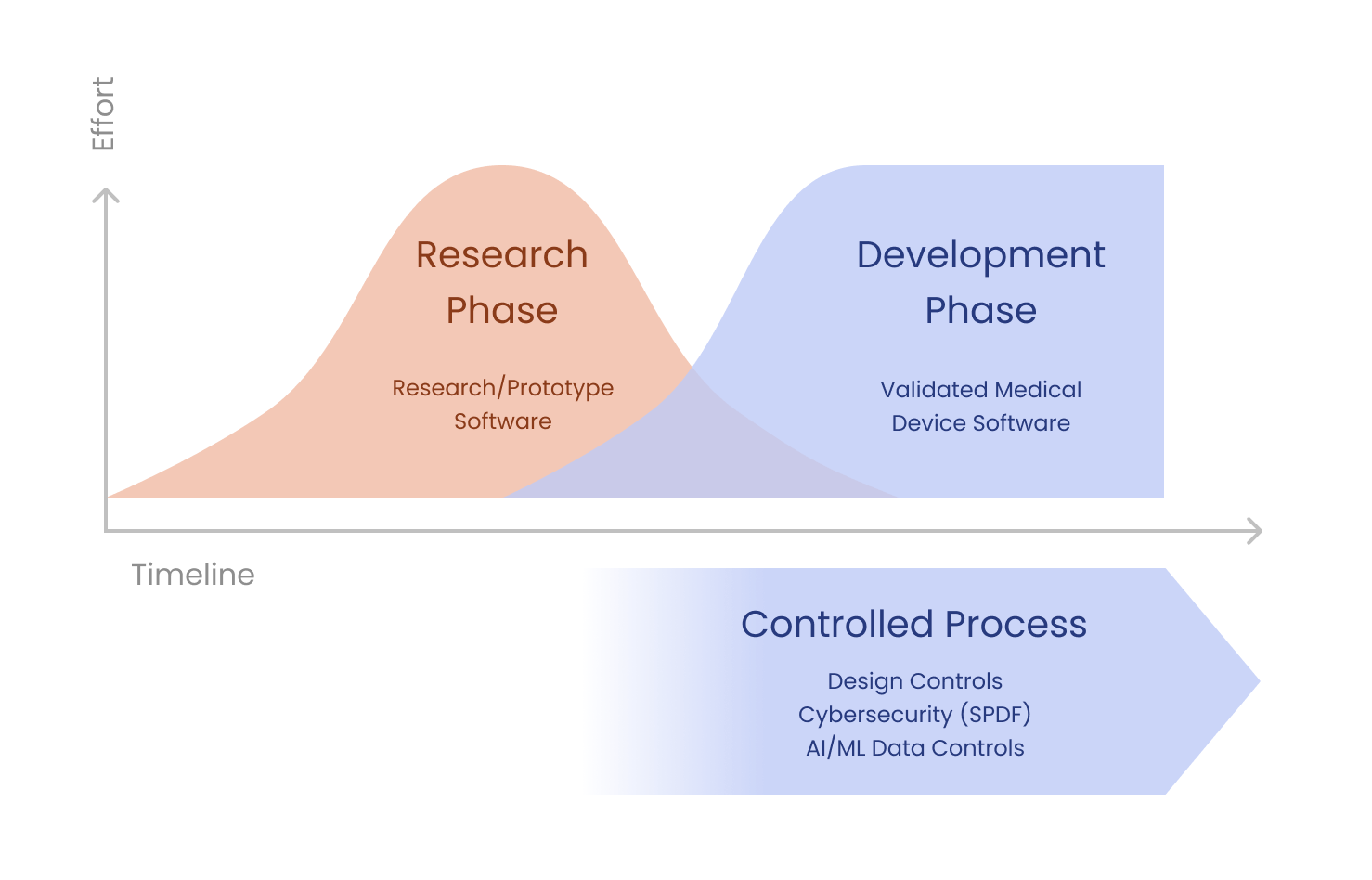
Vibe-coded prototypes are a great way to convey design intent. They are not the finished medical device, and they were never meant to be. They are the core artifact the regulated wrapper encases: the fastest way for clinicians to convey design intent, refine the intended use, and crystallize the requirements that the regulated build will then verify against.
A vibe-coded prototype is to a cleared medical device what:
- A whiteboard sketch is to a finalized engineering drawing.
- An MVP is to a productionized application.
- A research notebook is to a deployable algorithm.
- A LabVIEW prototype is to a Python rewrite.
The Vibe-to-Clearance Pipeline for AI Algorithms 🔗
![]()
If you have an AI-generated prototype that you want to bring to market, you cannot simply ship it as a medical device. You must adapt it. (The FDA never sees the code itself, only the documentation, risk file, and verification evidence that describe it. But that evidence has to actually be true of the code, which is where the work lives.) However, this adaptation is not a complete rewrite, as some consultants with less software development experience might tell you.
Instead, it is a structured process of careful review and remediation with the necessary regulatory evidence.
Deep Dive: The Anatomy of an AI-Generated Code Refactor 🔗
Let's walk through a hypothetical case study to illustrate exactly how a vibe-coded prototype is transformed into a compliant medical device.
Imagine a team of cardiologists who have developed a novel algorithm for detecting early signs of heart failure from wearable ECG data. They are not software engineers, but they used an LLM to "vibe code" a functional prototype in Python. The prototype works beautifully on their test dataset, but the code is a mess. It lacks error handling, uses unverified open-source libraries, and has zero documentation.
If they take this prototype to a traditional regulatory consultant, they will likely be told to throw it away and rewrite it from scratch. This would delay their time-to-market by a year or more, and every month of delay is a month where the predicate field shifts, competitors clear adjacent indications, and the clinical evidence base they were trying to advance moves on without them.
I suggest taking a different approach. I wrap the prototype. The vibe-coded artifact is treated as the captured design intent. The job is to encase it in a regulated implementation, not to throw it away.
Step 1: Establishing the Boundary 🔗
I start by treating the vibe-coded algorithm as a "black box." I work with the cardiologists to formally document the design intentions via the Software Requirements Specification (SRS). What exact data format does the algorithm expect? What exact output does it produce? What are the performance requirements (e.g., sensitivity, specificity, latency)?
Step 2: Risk Management (ISO 14971) 🔗
Next, I conduct a rigorous hazard analysis. What happens if the algorithm receives corrupted data? What happens if it produces a false positive? What happens if a cardiologist misinterprets the result? For each hazard, I define risk controls. Some of these controls will be implemented outside the algorithm (e.g., the wearable device must validate the signal quality before sending it). Other controls must be implemented within the software wrapper.
Step 3: The Wrapper Architecture 🔗
Instead of rewriting the algorithm, I use it as a starting point and build a wrapper around it that carries the prototype's design intent into a verifiable runtime. The wrapper ensures design invariants are currently met and will continue to be met:
- Input Validation: The wrapper checks every piece of incoming data to ensure it meets the required format and quality standards. If the data is bad, the wrapper rejects it before it ever reaches the vibe-coded algorithm.
- Error Handling: The wrapper monitors the execution of the algorithm. If the algorithm crashes or throws an exception, the wrapper catches it and fails gracefully, ensuring the system remains in a safe state.
- Output Verification: The wrapper checks the output of the algorithm to ensure it is within expected bounds before passing it on to the clinician.
Step 4: SOUP and SBOM 🔗
I then analyze the vibe-coded codebase to identify all third-party dependencies (Software of Unknown Provenance, or SOUP). I create a comprehensive Software Bill of Materials (SBOM) and assess each dependency for known cybersecurity vulnerabilities. If the LLM used an insecure or outdated library, I replace it with a secure alternative. If the LLM write multiple duplicate interfaces for the same functionality, this is where I will remediate that. This is where the SOUP gets un-SOUP-ified.
Step 5: Verification 🔗
Finally, I prove that the system works. I write an extensive suite of automated tests that exercise the wrapper and the algorithm. I test edge cases, boundary conditions, and failure modes. I generate the traceability matrix that links every requirement to a risk control and a passing test result.
The result is a fully compliant medical device. The core logic is still the original vibe-coded Python, but it is now encased in a fortress of verified, traceable engineering. The code stayed. The evidence was added.
AI-Generated SaMD and Good Machine Learning Practice (GMLP) 🔗
The FDA's AI/ML Action Plan also heavily emphasized the development of Good Machine Learning Practice (GMLP). In 2021, the FDA, Health Canada, and the UK's MHRA jointly published 10 guiding principles for GMLP [5]. These principles were later formalized by the International Medical Device Regulators Forum (IMDRF) in a document finalized in January 2025 [6].
GMLP is essentially the AI/ML equivalent of Good Manufacturing Practice (GMP) or Good Clinical Practice (GCP). It provides a framework for ensuring that AI models are developed, validated, and maintained in a safe and effective manner.
How does AI-generated code intersect with GMLP? At first glance, the two might seem fundamentally opposed. GMLP demands rigorous data governance, clear model architectures, and comprehensive performance testing. AI-assisted coding, by its very nature, is often exploratory and unstructured.
However, the two can be reconciled. GMLP does not dictate how the code is written; it dictates how the model is managed. If a vibe-coded prototype is used to train a machine learning model, the resulting model must still adhere to GMLP principles. This means:
- Multi-Disciplinary Expertise: The team developing the model must include domain experts (e.g., clinicians) and data scientists. AI-assisted coding actually facilitates this by allowing clinicians to directly interact with the code generation process.
- Good Software Engineering and Security Practices: This is where the "wrapper" approach I discussed earlier becomes critical. The vibe-coded model must be integrated into a secure, well-engineered software system.
- Clinical Study Participants and Data Sets: The data used to train and test the model must be representative of the intended patient population. AI-generated code does not change this requirement.
- Training Data Sets Are Independent of Test Sets: This is a fundamental principle of machine learning that must be strictly enforced, regardless of how the code was generated.
- Selected Reference Datasets Are Based Upon Best Available Methods: The ground truth used to evaluate the model must be reliable.
- Model Design Is Tailored to the Available Data and Reflects the Intended Use: The architecture of the model must be appropriate for the task.
- Focus Is Placed on the Performance of the Human-AI Team: If the model is intended to assist a clinician, the performance of the combined human-AI system must be evaluated.
- Testing Demonstrates Device Performance During Clinically Relevant Conditions: The model must be tested in realistic scenarios.
- Users Are Provided Clear, Essential Information: The labeling and instructions for use must clearly explain the model's capabilities and limitations.
- Deployed Models Are Monitored for Performance and Re-training Risks are Managed: The manufacturer must have a plan for monitoring the model's performance in the real world and managing the risks associated with retraining.
By adhering to these principles, a manufacturer can ensure that a vibe-coded AI/ML model is safe and effective, even if the underlying code was generated by an LLM.
The Cybersecurity Imperative 🔗
One of the most valid criticisms of AI-generated code is its potential impact on cybersecurity. LLMs are trained on vast amounts of open-source code, much of which contains known vulnerabilities or insecure coding patterns. If an engineer blindly accepts LLM-generated code, they may inadvertently introduce these vulnerabilities into their medical device.
The FDA has become increasingly stringent regarding cybersecurity in recent years. In 2023, the agency gained new statutory authority to require cybersecurity information in premarket submissions, including a Software Bill of Materials (SBOM) and a plan for monitoring and addressing postmarket cybersecurity vulnerabilities [7].
How can an AI-generated medical device meet these stringent requirements?
The answer lies in rigorous, automated security testing and dependency management. When an AI-generated codebase is transitioned to a compliant medical device, the following cybersecurity practices must be implemented:
- Static Application Security Testing (SAST): The vibe-coded codebase must be scanned using SAST tools to identify insecure coding patterns, hardcoded secrets, and other vulnerabilities. Any issues identified must be remediated before the code is deployed.
- Dynamic Application Security Testing (DAST): The running application must be tested using DAST tools to identify vulnerabilities that only manifest during execution, such as injection flaws or cross-site scripting (XSS).
- Software Composition Analysis (SCA): As mentioned earlier, a comprehensive SBOM must be generated using SCA tools. This SBOM must list all third-party dependencies (SOUP) used by the vibe-coded application, along with their versions and known vulnerabilities.
- Vulnerability Management: The manufacturer must establish a process for continuously monitoring the SBOM for new vulnerabilities and deploying patches or mitigations as needed.
- Threat Modeling: A formal threat model must be developed to identify potential attack vectors and ensure that appropriate security controls are in place.
By implementing these practices, a manufacturer can mitigate the cybersecurity risks associated with AI-generated code and demonstrate to the FDA that their device is secure. The LLM may have written the code, but the manufacturer is responsible for securing it.
Agentic AI Best Practices 🔗
If an LLM is holding the pen, whether through Cursor, Claude Code, ChatGPT Codex, Lovable, Replit, Antigravity, GitHub Copilot, Windsurf, Devin, Aider, or whatever tool ships next quarter, the discipline shifts. The tool changes; the regulatory constraints do not. What used to be caught in ad-hoc, end-of-pipeline code review now has to be enforced upstream through specification, structure, and acceptance criteria, with the review itself promoted from optional polish to a defined acceptance step (see #6). The agent will write whatever you let it write. Your job is to make "whatever" small, predictable, bounded, and verifiable.
Six practices we use at Innolitics when building FDA-regulated software with agentic AI:
1. Specification-Driven Development 🔗
The specification is the source of truth. Not the code. Not the prompt. The spec.
This is exquisitely important given the memory limitations of AI agents. They forget everything about the codebase on every new coding session so must be quickly brought back up to speed on the product, design patterns, requirements, and everything else that human engineers carry around in their heads every day.
2. Clean, Simple, Concise Code With No Surprises 🔗
Boring code is good code.
Every function should do exactly what its name says: no hidden side effects, no implicit state, no exception handling that swallows context. If the next engineer has to read the body to understand the behavior, the function failed.
3. Self-Documenting Interfaces 🔗
Interfaces should not need a manual. Type signatures, parameter names, and return contracts encode the intent. If you need a comment to explain what a function does, the function is wrong.
This matters more in agentic workflows than in human-authored code. The next agent that touches the codebase reads interfaces, not your design rationale doc. The next engineer who inherits the module reads the same thing. A self-documenting interface is the contract that survives across iterations and across teammates.
4. Keep It Stupidly Simple 🔗
KISS just got existentially important in the age of agentic AI software engineering. Complex code breeds more complex code. It is a positive feedback loop that results in a meltdown. I described this in this article https://innolitics.com/articles/ai-native-engineering-transformation/
5. Lines-of-Code and Complexity Budgets in the Specification 🔗
Set hard ceilings in the spec. Maximum function length. Maximum cyclomatic complexity. Maximum module size.
Picture this. You are a manager delegating a task to a junior engineer. The engineer goes off and comes back with a 1k+ line diff for something that should have been a 1 liner. Had you specified this upfront, the engineer would have likely found the right solution more autonomously. If indeed the problem is genuinely more complicated than a one line fix, the engineer would have more likely raised the concern earlier on and you can decide appropriately to descope problematic features.
6. Acceptance Criteria That Include Code Review 🔗
Generated code is not done when it passes tests. It is done when the diff has been reviewed against the acceptance criteria and signed off.
Some acceptance criteria can be verified through browser automation, unit tests, integration tests, or code review.
The Role of the Regulatory Consultant in the Age of AI-Generated Code 🔗
As the software engineering paradigm shifts toward AI-generated code, the role of the regulatory consultant must also evolve. Historically, regulatory consultants have often acted as gatekeepers, enforcing strict adherence to traditional software development life cycles (SDLCs). They have been trained to look for comprehensive design documents, detailed code reviews, and manual traceability matrices.
When presented with an AI-generated codebase, a traditional consultant's instinct is often to reject it. They see the lack of human-authored code and the reliance on probabilistic LLMs as insurmountable regulatory hurdles.
This approach is no longer viable. Regulatory consultants must adapt to the new reality of AI-assisted software development. They must become facilitators rather than gatekeepers.
A modern regulatory consultant must understand how to apply the principles of IEC 62304 and ISO 14971 to an AI-generated codebase. They must be able to guide manufacturers through the process of building the "wrapper", establishing the boundaries, conducting the hazard analysis, and implementing the necessary risk controls.
They must also be well-versed in the FDA's latest guidance on AI/ML, including the use of PCCPs and the application of Good Machine Learning Practice (GMLP). They must be able to help manufacturers navigate the complex intersection of AI, cybersecurity, and medical device regulation.
At Innolitics, we have embraced this evolving role. We don't tell my clients to throw away their vibe-coded prototypes. We help them understand the regulatory landscape, identify the gaps in their evidence, and build the engineering pipelines necessary to achieve compliance. We believe regulatory expertise should enable innovation, not stifle it.
The Future of Medical Device Software Engineering 🔗
The medical device industry is notoriously slow to adopt new technologies. This caution is understandable; when software fails in a medical device, the consequences can be catastrophic. However, this caution can also stifle innovation and delay the delivery of life-saving technologies to patients.
AI-assisted coding represents a massive leap forward in software engineering productivity. It has the potential to democratize the development of medical devices, allowing clinicians, researchers, and domain experts to rapidly prototype and iterate on new ideas.
If the industry rejects AI-generated code outright, it will miss out on this incredible potential. Instead, the industry must embrace AI-assisted development while simultaneously adapting its regulatory and engineering practices to manage the associated risks.
This means shifting the focus from how the code is written to how the code is verified and validated. It means embracing automated testing, rigorous risk management, and comprehensive cybersecurity practices. It means recognizing that compliance is not a property of the code itself, but a property of the evidence surrounding the code.
The FDA has already demonstrated its willingness to adapt to new software paradigms, from the Agile process to deployment methods like CLI, SDK, and containerization. The agency's frameworks, including substantial equivalence, the PCCP, and GMLP, are flexible enough to accommodate a new methodology for the agentic era.
Those who take calculated risks will be able to bring innovative medical devices to market faster than ever before. Those that take too many risks will end up getting warning letters and get shut down by FDA. Those who don’t take enough risks will be left behind, clinging to outdated development paradigms while their competitors embrace the future.
Bring Your AI-Generated Code to Specification 🔗
The FDA is ready to accept AI assisted development methodologies in medical devices, provided they are created with a compliant development process in mind. The regulatory frameworks that absorbed the Agile method, SCRUM, CLI, the SDK, Docker, and the cloud are fully capable of absorbing devices built with LLM-generated code. The paradigm itself is acceptable; the FDA never receives the code, only the documentation, traceability, and verification evidence that describe it.
At Innolitics, we specialize in bridging the gap between modern software engineering and FDA compliance. My team of engineers and regulatory experts can help you adapt your prototype into an IEC 62304-compliant, traceable codebase with full verification.
- Software Development Services: We can refactor your prototype, implement robust testing, generate the necessary software documentation, and turn it into a shippable device. Learn more about our End-to-End SaMD Development.
- FDA Regulatory Consulting: We can guide you through risk classification, predicate selection, and 510(k) authoring.
- Guaranteed Clearance: We are so confident in our process that we offer Guaranteed AI/ML SaMD 510(k) Clearance.
- Cybersecurity: We can secure your vibe-coded codebase, ensuring SBOM compliance and dependency hygiene.
References 🔗
[1] Karpathy, A. (2025, February 6). There's a new kind of coding I call 'vibe coding'...X (formerly Twitter).
[2] International Electrotechnical Commission. (2006).IEC 62304: Medical device software - Software life cycle processes.
[3] International Organization for Standardization. (2019).ISO 14971: Medical devices - Application of risk management to medical devices.
[4] U.S. Food and Drug Administration. (2021).Artificial Intelligence/Machine Learning (AI/ML)-Based Software as a Medical Device (SaMD) Action Plan.
[5] U.S. Food and Drug Administration, Health Canada, and Medicines and Healthcare products Regulatory Agency. (2021).Good Machine Learning Practice for Medical Device Development: Guiding Principles.
[6] International Medical Device Regulators Forum. (2025).Good Machine Learning Practice for Medical Device Development: Guiding Principles.
[7] U.S. Food and Drug Administration. (2023).Cybersecurity in Medical Devices: Quality System Considerations and Content of Premarket Submissions.