Through April 2026, the FDA has authorized fifty-one AI/ML-flagged devices in pathology-relevant review panels, but only seven of those algorithms actually analyze whole-slide images; the remaining forty-four are hematology analyzers, molecular IVDs, and cytology systems, and that composition tells a more useful story than the headline number does. The structural fact most newcomers miss is that a pure-software pathology algorithm — the kind a startup with ten ML engineers and zero wet-lab reagents might ship — is still reviewed as an In Vitro Diagnostic in OHT7 rather than as Software as a Medical Device in OHT8, which translates into a different review office, a different predicate ecosystem, and a meaningfully different evidence bar. If your mental model for "ship an AI medical device" came from radiology, you will prepare the wrong submission.

What "Pathology AI" Actually Means in the 2026 Dataset 🔗
The FDA does not publish a "pathology AI" bucket. The 51 devices here are every AI/ML-flagged authorization across pathology-adjacent panels (Pathology, Medical Genetics, Hematology, Microbiology, Immunology) in Innolitics' Device Explorer. The flag is applied to records whose summary, intended use, or indications describe trained classifiers or ML-based decision-support. Range: CellaVision DiffMaster Octavia (2001) through Checkcells' Seaman Pro (April 2026).
There are two common distortions worth avoiding when counting this way: WSI-only counts produce single-digit totals and miss the analyzers that are actually moving specimens through labs, while "everything automated" counts sweep in decades of rules-based hematology and inflate the number beyond recognition. The AI/ML flag splits the difference by keeping modern trained-classifier systems (CellaVision DM-series, Scopio's full-field smear platforms, Sight OLO, the genomic profiling pipelines whose variant calling and signature scoring are ML-based) while excluding the rules-based legacy that long predated machine learning.
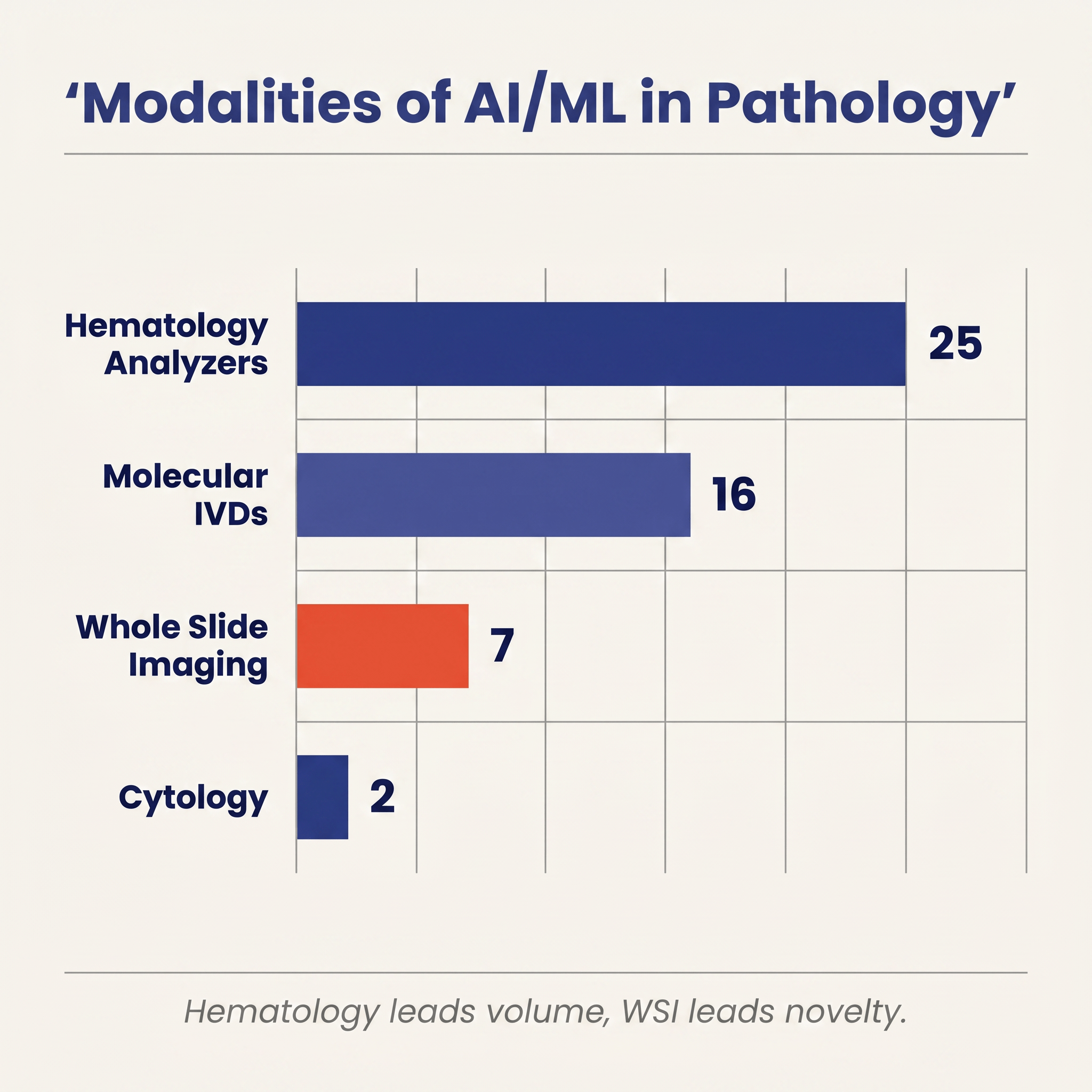
Inside that set, four modalities emerge and they are wildly uneven in both count and clinical footprint.
Hematology and body-fluid analyzers dominate by volume, with twenty-five authorizations. This is the category that has been quietly using machine learning in production the longest. CellaVision's automated white-blood-cell classifier has been iterating on the FDA since 2001. Scopio Labs' full-field peripheral blood smear system, Pixcell's HemoScreen, Sight Diagnostics' OLO, Athelas Home, Sysmex's XR series, and semen-quality analyzers from Bonraybio and Checkcells all live here. These devices are not the romantic version of pathology AI (no prostate cancer detection on a gigapixel slide), but they are the version that has actually put AI-assisted morphology into routine clinical workflows across thousands of labs. Both of 2026's pathology-relevant clearances to date are in this bucket: Athelas Home (K243348, February 6) and Checkcells' Seaman Pro (K252228, April 9).
Molecular IVDs contribute sixteen authorizations and are the other large cohort. This is Foundation Medicine's FoundationOne CDx, Myriad's myChoice HRD CDx, Exact Sciences' Cologuard Plus, Guardant's Shield, Tempus' xT and xR, Agendia's MammaPrint, Nanostring's Prosigna, and recent additions such as Biocartis' Idylla CDx MSI Test (P250005) and Tempus AI's xR IVD (K241868). The pathology here is genomic rather than morphologic: the assay performs sequencing or hybridization in a lab, and the "AI" is the machine-learning model that turns raw genomic reads into clinically actionable calls: variants, tumor mutational burden, homologous recombination deficiency scores, tissue-of-origin predictions, MSI status, and methylation-based screening results.
Whole-slide imaging AI, the category most clinicians and journalists mean when they say "digital pathology AI," contributes just seven authorizations. Three of them (Paige Prostate, Ibex's Galen Second Read, ArteraAI Prostate) are the contemporary wave of computational pathology. The other four are legacy IHC scoring systems from Applied Imaging (×2), Tripath/Ventana, and Aperio, dating 2004–2009 under older product codes (NOT, NQN). Seven devices in twenty-plus years is the true scale of cleared WSI AI.
Cytology rounds out the count with two authorizations: Becton Dickinson's BD MAX CTGCTV2 System (K182692) and Hologic's Genius Digital Diagnostics System with Genius Cervical AI (DEN210035), which assists cytotechnologists in interpreting ThinPrep Pap tests.
The visibility disparity across these four modalities is instructive in itself: hematology dominates the volume column but is so thoroughly embedded in lab workflows that few people outside the hematology community think of it as "AI" at all, while WSI AI leads on press coverage and academic visibility despite being numerically tiny, and molecular IVDs sit between the two with substantial count, large clinical and commercial impact, and an almost-default IVD classification driven by the fact that the underlying assay is physical even when the decision-making is algorithmic.
The Arc From 1999 to April 2026 🔗
Twenty-two authorizations accumulated across the first two decades (1999 through 2019), driven almost entirely by hematology analyzers and a handful of molecular tests; from 2020 through 2023 the pace settled into a steady two-to-four-per-year rhythm; 2024 held that cadence at four authorizations, including the cytology landmark with Hologic's Genius Cervical AI in January; and then 2025 more than doubled the prior year's total with ten authorizations in a single calendar year.
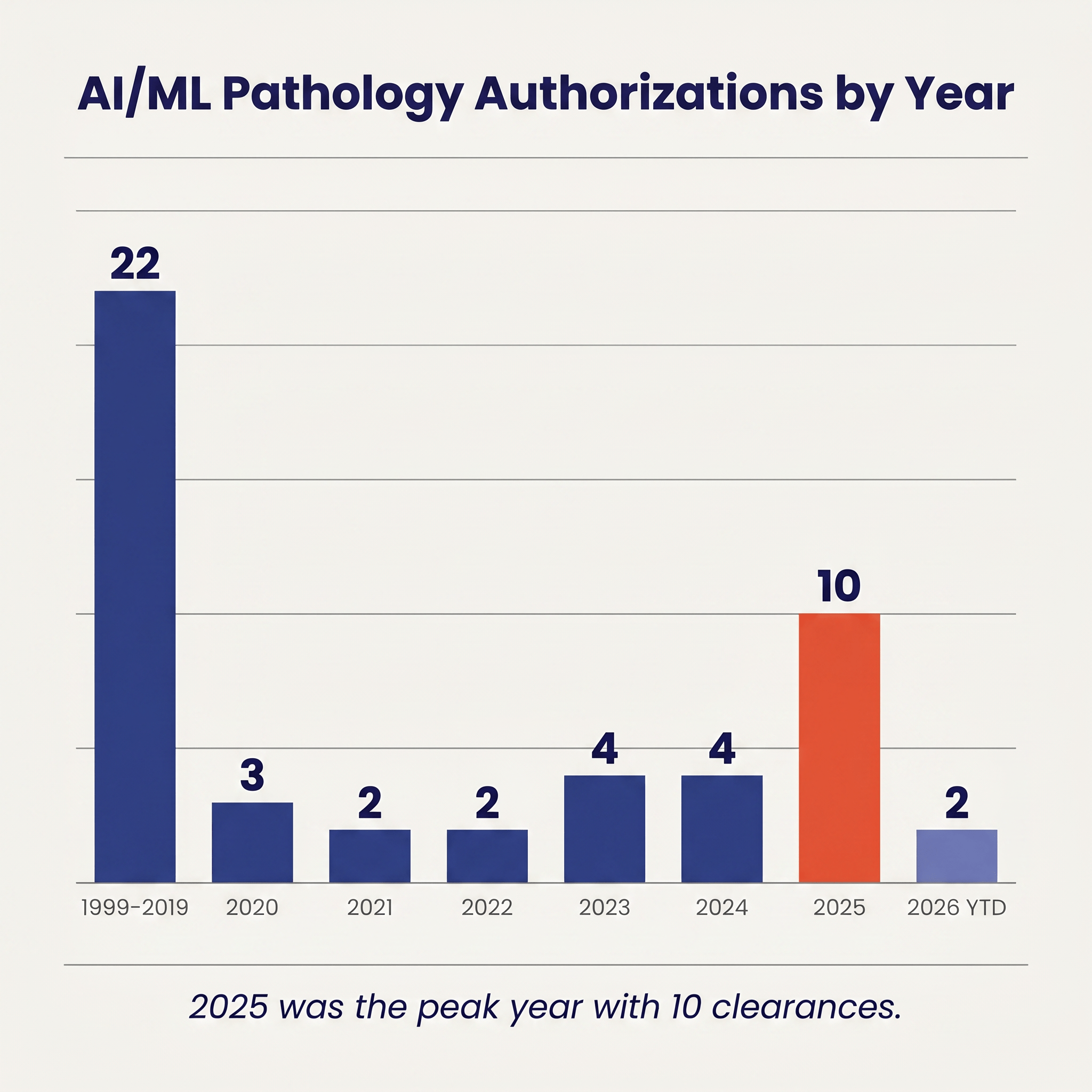
2025's ten authorizations included the first QPN 510(k) clearance (Ibex Galen Second Read, K241232), the first prognostic WSI De Novo (ArteraAI Prostate, DEN240068), two PZM molecular IVDs (Tempus xR IVD, K241868, and Geneseeq's GENESEEQPRIME, K250003), a PMA for Biocartis' Idylla CDx MSI (P250005), and four hematology and semen-analyzer 510(k)s spread across the second half of the year.
2026 has opened quietly by comparison, with only two hematology clearances through April 28 (Athelas Home, K243348, and Checkcells' Seaman Pro, K252228) and no new WSI, cytology, or molecular IVD authorizations to date. Four months is a small sample — especially in a category whose 2025 milestones clustered in the second half of the year — so the early-2026 cadence is worth treating as a data point rather than as a trend.
Three Pathways, and Why De Novo Does the Heavy Lifting 🔗
Across the fifty-one devices, the three regulatory pathways split in a specific way: forty-one 510(k) clearances, seven PMAs, and three De Novos.

That distribution is heavily shaped by category. The 510(k) column is dominated by hematology analyzers and legacy molecular tests, all of which have decades-old predicates to claim substantial equivalence against. The PMA column is mostly companion diagnostics, including FoundationOne CDx, Tempus xT CDx, myChoice HRD CDx, Cologuard Plus, Guardant Shield, 4Kscore, and Biocartis' Idylla CDx MSI, where the FDA has required full premarket approval because the test is tied to a specific therapeutic decision.
The three De Novos, namely Paige Prostate (DEN200080, 2021), Hologic Genius Cervical AI (DEN210035, 2024), and ArteraAI Prostate (DEN240068, 2025), are where the regulatory architecture for modern pathology AI has actually been built. Each of them established a new product code, which means each of them simultaneously cleared itself and created a regulatory slot that future devices can use as a predicate under 510(k). QPN was created by Paige Prostate and has now been used as the predicate for Ibex's Galen Second Read. QYV was created by Genius Cervical AI. SFH was created by ArteraAI Prostate. In a category with almost no legacy predicates (WSI AI has no 1990s ancestor to be substantially equivalent to), De Novos are not a minority pathway. They are the scaffolding that lets the 510(k) pathway work at all.

Seen another way, the top product codes in the dataset trace the history of how pathology AI has actually been reviewed. JOY (automated hematology differential cell counter) carries eleven authorizations. GKZ (hematology analyzer) carries eight. POV (semen analyzer) carries five. OIW (tissue-of-origin molecular test) carries four. NOT and NYI each carry three. The modern WSI AI codes (QPN, QYV, SFH) each have only one or two members, because they did not exist until 2021 onward. This is what it looks like for a new regulatory category to be built from scratch inside the existing taxonomy.
The Paradigm: Why Software-Only Pathology AI Is Still an IVD 🔗
Paige Prostate's De Novo classified the device under Product Code QPN in 21 CFR 864.3700, a regulation that sits inside Part 864 (Hematology and Pathology Devices) and therefore inside the IVD framework rather than the SaMD framework. The same is true for Hologic's Genius Cervical AI under QYV, ArteraAI Prostate under SFH, and Ibex's Galen Second Read riding QPN as its predicate: every modern software-only pathology algorithm in the dataset has been reviewed in OHT7 rather than in OHT8, even though the products themselves look, from a software-engineering standpoint, almost identical to a radiology SaMD.

In radiology, an algorithm that reads an X-ray, a CT, an MRI, or an ultrasound lives in SaMD land. The review office, the guidance documents, the predicate ecosystem, and the clinical validation conventions all assume the product is a piece of software whose input happens to be an image acquired by a piece of radiological imaging equipment regulated separately. In pathology, that separation does not hold. The FDA's definition of an IVD is broad: reagents, instruments, and systems intended for use in the diagnosis of disease or other conditions using specimens taken from the human body. A digitized whole-slide image is treated as a digital representation of a physical tissue specimen, and the software that interprets it is treated as an extension of the in vitro diagnostic process that begins with the biopsy and continues through fixation, staining, scanning, and interpretation.
All four modern WSI AI authorizations sit under Part 864, and the framing is not theoretical: it shows up in the decision letters, in the way scanner and stain constraints get baked into the label, and in how Ibex's Galen Second Read had to inherit the boundaries Paige had already established in order to come through as a 510(k) at all.
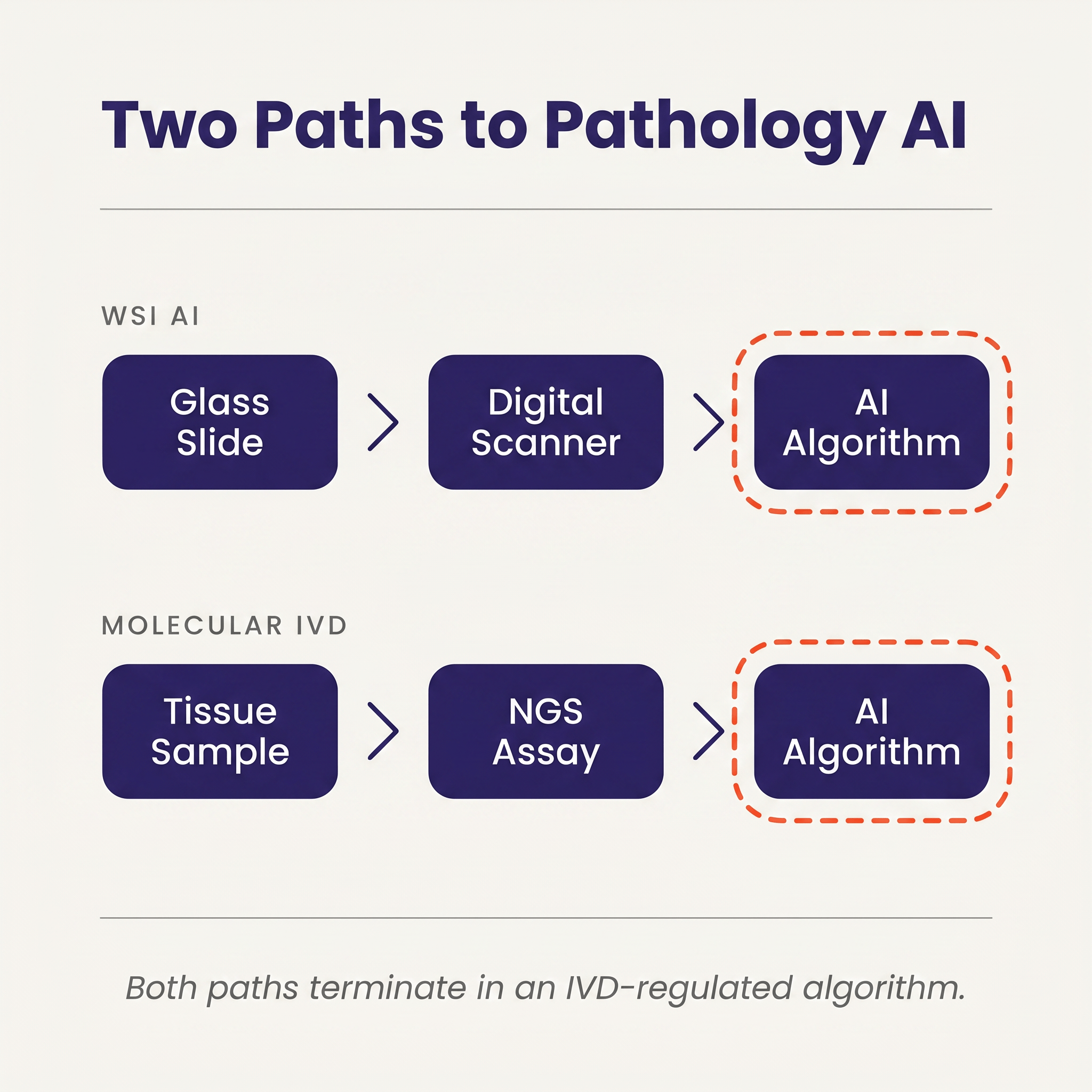
The consequence is that two seemingly different products, a WSI AI algorithm from a pure-software company and a molecular IVD from a sequencing company, end up in the same regulatory bucket. Both paths start with a physical specimen. Both involve an instrument-mediated transformation of that specimen into a digital representation (scanner or sequencer). Both end with an algorithm that turns that digital representation into a diagnostic call. From the agency's perspective, both are IVDs and both are evaluated against IVD evidentiary norms.
This has downstream effects that software-only developers entering pathology often underestimate.
Clinical validation is specimen-aware, not just model-aware. OHT7 reviewers do not evaluate an algorithm the way an OHT8 reviewer evaluates a radiology SaMD. They have to account for more variability in the specimen to clinician pipeline: the types of stains used, the scanners that produced the training and validation images, the specific lab protocols that the studies ran on, the reader variability among the pathologists who produced the ground truth, and the lot-to-lot and site-to-site variation in the upstream physical specimen. Paige Prostate's authorization, for example, is specific about which scanners it is cleared on and under what magnification and staining conditions. A label claim extended to a new scanner typically requires additional validation.
Post-market surveillance looks different. Under OHT7, post-market expectations focus on analytical drift, assay-level adverse events, and lot-traceable corrections. For software that can silently shift as scanners and stains evolve in the field, this is a meaningful operational cost, not a paperwork exercise.
The lesson is simple and easy to miss: the agency's framing treats your algorithm as a component of a diagnostic assay rather than as a standalone software product that happens to read a medical image, and the submission has to be designed around that framing from the first scoping conversation rather than retrofitted to it after a Pre-Sub goes sideways.
The Scale Gap Between Radiology AI and Pathology AI 🔗

The math is worth holding in mind whenever anyone talks about "AI in healthcare" as a single market: cardiology alone cleared roughly ninety AI/ML devices in 2025, while the pathology WSI AI column on the same FDA list contains seven authorizations cumulative, spread across more than two decades. Several structural reasons drive that gap, and they compound rather than acting in isolation.
Data standardization. DICOM has been the lingua franca of radiology images for decades. Pathology's equivalent standard, DICOM WSI, is technically mature but adoption is still partial, and the de facto file formats are proprietary, including Aperio SVS, Hamamatsu NDPI, Philips iSyntax, 3DHistech MRXS, Leica GT450 SCN, and so on. A radiology startup can train on open datasets aggregated across hospitals with modest engineering. A pathology startup has to build format-conversion and color-normalization tooling before it can train anything, and must document that pipeline for the FDA.
Infrastructure penetration. Radiology departments went fully digital by the mid-2000s. Pathology remains largely glass. Even in 2026, after years of vendor investment and pandemic-accelerated digital pathology deployments, most U.S. labs still use glass slides and optical microscopes for primary diagnosis, with WSI reserved for consults, education, and selected computational use cases A cleared WSI AI algorithm has a smaller addressable installed base than a cleared radiology SaMD.
Regulatory burden. The IVD framing described above raises the evidentiary cost per submission compared to a typical radiology CADe/CADx, where cleared software predicates are abundant and the review conventions are well worn. For pure-software startups with limited wet-lab infrastructure, this often translates into expensive multi-site clinical studies that would not be required of an analogous radiology product.
Reference-standard cost. Ground truth for radiology AI often comes from existing reads, existing reports, and retrospective cohorts. Ground truth for pathology AI frequently requires expert pathologist re-review of large slide sets, sometimes multiple pathologists per slide to capture inter-reader variability. That is slow and expensive in a way that is not comparable to collecting radiology labels.
Taken together, these factors help explain not only the quantitative gap but also why the gap has not closed quickly despite obvious clinical demand and active venture-backed competition.
The Innovators Actually Clearing the IVD Bar 🔗
The list of companies that have actually brought computational pathology through the FDA is short, concrete, and worth looking at directly rather than through the abstraction of product codes.
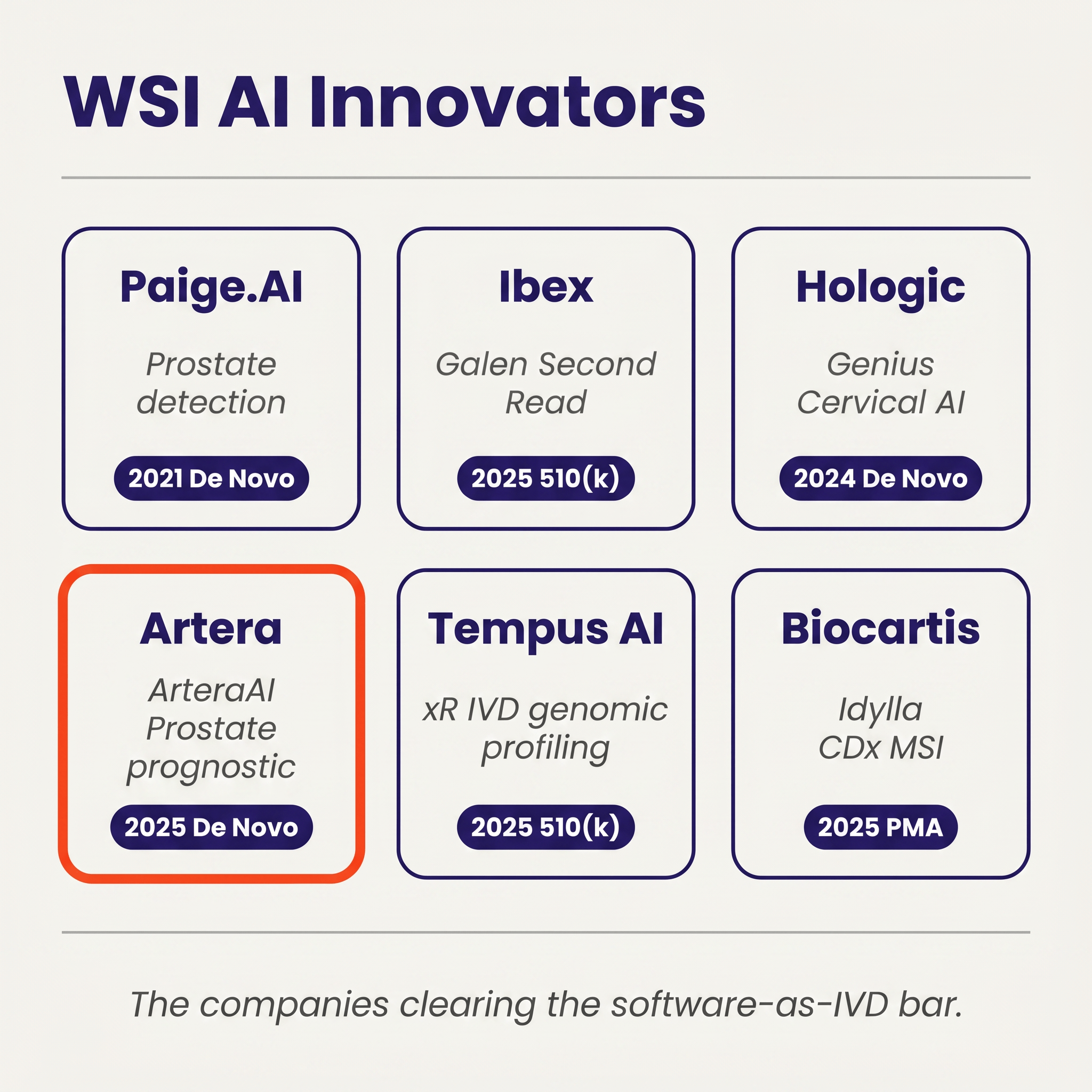
Paige.AI (Paige Prostate, DEN200080, September 2021) created QPN as the first product code for WSI cancer detection, and its decision summary made the software-as-IVD framing explicit while locking in the scanner and stain constraints that every QPN follow-on now inherits.
Ibex Medical Analytics (Galen Second Read, K241232, January 2025) became the first 510(k) to clear under QPN, which is the proof point for the broader pathway logic: once a De Novo establishes a regulatory slot, subsequent entrants can come in through substantial equivalence as long as they respect the predicate's scanner-and-stain envelope and match its performance.
Hologic (Genius Digital Diagnostics System with Genius Cervical AI, DEN210035, January 2024) did for cytology what Paige had done for histology, creating QYV for cervical cytology AI; because cervical screening is the highest-volume routine workflow in the dataset, QYV plausibly carries the broadest near-term adoption ceiling of the three modern WSI/cytology codes.
Artera (ArteraAI Prostate, DEN240068, July 2025) created SFH and is the first FDA-authorized prognostic WSI AI device, predicting androgen-deprivation-therapy benefit from a biopsy slide combined with clinical data and, in doing so, opening a prognostic/predictive lane that has, until now, lived almost entirely in academic publications.
Tempus AI (xR IVD, K241868, September 2025) and Biocartis (Idylla CDx MSI Test, P250005, August 2025) anchor the modern molecular side, with an RUO-to-IVD genomic profiling panel cleared under PZM and a PMA companion diagnostic for microsatellite instability detection, respectively.
What the Snapshot Suggests About the Next Two Years 🔗
As of April 28, 2026, three patterns seem worth watching.
The De Novo scaffolding is in place for three sub-domains, but only QPN has been tested as a predicate. QPN has produced one follow-on 510(k) so far (Ibex Galen Second Read), while QYV and SFH have not yet been used as predicates by any cleared device, which means QPN-adjacent submissions should plausibly clear faster than the De Novo timeline going forward, but it remains an open question whether adjacent sub-domains (IHC quantification, broader biomarker prediction, non-prostate cancers) will be able to use the existing codes or will require fresh De Novos of their own.
Molecular IVDs will continue to dominate the pathology AI volume story even as WSI gets more press. Six of the ten 2025 authorizations, and the two PMA-class decisions, came from molecular IVD developers. This is the part of pathology AI that is already paid for by reimbursement and already integrated into oncology treatment decisions. Expect this column to keep growing regardless of how quickly WSI adoption progresses.
The quiet 2026 start is a data point, not a trend yet. Two hematology clearances through mid-April, after ten authorizations in 2025, could reflect nothing more than post-peak queue dynamics. But it is also a useful reminder that pathology AI's cadence is driven by a small number of active developers, and a single quarter without a major WSI or cytology submission can visibly change the annual curve in a category this small.
Pathology AI is now a real category supported by fifty-one cleared devices, three established modern product codes, and a small but identifiable cohort of active innovators that have actually shipped through OHT7. The math is still humbling, though, because a single radiology specialty (cardiology) cleared roughly ninety AI/ML devices in 2025 alone while pathology has produced fifty-one across more than two decades, and the binding constraint on closing that gap is not developer pace but the combined weight of data standardization, lab digitization, and IVD-grade evidence requirements that pathology AI has to clear before any of those developers can ship.
References 🔗
[1] FDA Device Explorer by Innolitics. Authorization records for AI/ML-flagged pathology-relevant devices (panels PA, Pathology, Medical Genetics, HE, MI, Immunology; product codes QPN, QYV, SFH, QRF, OIW, NOT, NQN, NYI, PQP, PJG, PHP, PZM, SFL, JOY, GKZ, POV, GKF, OUY, PSY, QKQ). Data retrieved April 28, 2026. https://fda.innolitics.com
[2] Paige Prostate De Novo decision letter, DEN200080 (September 21, 2021). https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfpmn/denovo.cfm
[3] Hologic Genius Cervical AI De Novo decision letter, DEN210035 (January 31, 2024). https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfpmn/denovo.cfm
[4] ArteraAI Prostate De Novo decision letter, DEN240068 (July 31, 2025). https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfpmn/denovo.cfm
[5] Ibex Galen Second Read 510(k) summary, K241232 (January 24, 2025). https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfpmn/pmn.cfm
[6] Tempus AI xR IVD 510(k) summary, K241868 (September 19, 2025). https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfpmn/pmn.cfm
[7] Biocartis Idylla CDx MSI Test PMA approval, P250005 (August 15, 2025). https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfpma/pma.cfm
Appendix
| record_id | decision_date | decision_year | pathway | product_code | review_panel | modality | applicant | device_name |
| K983551 | 5/27/1999 | 1999 | 510(k) | GKF | HE | hematology | In-Line Diagnostics Corp. | CRIT-SCAN II MONITOR |
| K003301 | 3/15/2001 | 2001 | 510(k) | JOY | HE | hematology | CellaVision AB | DIFFMASTER OCTAVIA AUTOMATIC HEMATOLOGY ANALYZER |
| K031715 | 1/8/2004 | 2004 | 510(k) | NOT | PA | wsi_ai | Applied Imaging Corp. | ARIOL HER-2/NEU IHC |
| K033840 | 2/10/2004 | 2004 | 510(k) | JOY | HE | hematology | CellaVision AB | CELLAVISION DM96 AUTOMATIC HEMATOLOGY ANALYZER |
| K033200 | 3/18/2004 | 2004 | 510(k) | NQN | PA | wsi_ai | Applied Imaging Corp. | ARIOL |
| K051282 | 8/16/2005 | 2005 | 510(k) | NOT | PA | wsi_ai | Tripath Imaging, Inc. | VENTANA IMAGE ANALYSIS SYSTEM - HER2/NEU |
| K070675 | 6/22/2007 | 2007 | 510(k) | NYI | PA | molecular_ivd | Agendia BV | MAMMAPRINT |
| K080252 | 7/21/2008 | 2008 | 510(k) | NYI | PA | molecular_ivd | Agendia BV | MODIFICATION TO MAMMAPRINT |
| K080896 | 7/30/2008 | 2008 | 510(k) | OIW | PA | molecular_ivd | Pathwork Diagnostics, Inc | PATHWORK DIAGNOSTICS TISSUE OF ORIGIN TEST |
| K080595 | 12/5/2008 | 2008 | 510(k) | JOY | HE | hematology | CellaVision AB | CELLAVISION DM96 WITH THE BODY FLUID APPLICATION |
| K080564 | 8/14/2009 | 2009 | 510(k) | NOT | PA | wsi_ai | Aperio Technologies | SCANSCOPE XT SYSTEM |
| K092116 | 5/12/2010 | 2010 | 510(k) | JOY | HE | hematology | Medica Corp. | EASYCELL CELL LOCATOR |
| K092967 | 6/8/2010 | 2010 | 510(k) | OIW | PA | molecular_ivd | Pathwork Diagnostics | PATHWORK TISSUE OF ORIGIN TEST KIT-FFPE (ORIGIN TE |
| K102778 | 9/16/2011 | 2011 | 510(k) | JOY | HE | hematology | CellaVision AB | CELLAVISION DM1200 WITH THE BODY FLUID APPLICTION |
| K120489 | 5/17/2012 | 2012 | 510(k) | OIW | PA | molecular_ivd | Pathwork Diagnostics, Inc | TISSUE OF ORIGIN TEST KIT FFPE |
| K141771 | 11/7/2014 | 2014 | 510(k) | NYI | PA | molecular_ivd | Nanostring Technologies, | PROSIGNA BREAST CANCER PROGNOSTIC GENE SIGNATURE A |
| K171315 | 8/1/2017 | 2017 | 510(k) | JOY | HE | hematology | CellaVision AB | Advanced RBC Application |
| P170019 | 11/30/2017 | 2017 | PMA | PQP | Pathology | molecular_ivd | Foundation Medicine, Inc. | FoundationOne CDx |
| K173839 | 3/15/2018 | 2018 | 510(k) | OIW | PA | molecular_ivd | Cancer Genetics, Inc. | Tissue of Origin Test Kit-FFPE |
| K180020 | 10/29/2018 | 2018 | 510(k) | GKZ | HE | hematology | Pixcell Medical Technolog | HemoScreen Hematology Analyzer |
| K180343 | 11/16/2018 | 2018 | 510(k) | POV | HE | hematology | Bonraybio Co., Ltd. | LensHooke X1 Pro Semen Quality Analyzer, LensHooke |
| K182692 | 1/8/2019 | 2019 | 510(k) | OUY | MI | cytology | Becton, Dickinson and Com | BD MAX CTGCTV2, BD MAX System |
| P190014 | 10/23/2019 | 2019 | PMA | PJG | Pathology | molecular_ivd | Myriad Genetic Laboratori | myChoice HRD CDx |
| K190898 | 11/1/2019 | 2019 | 510(k) | GKZ | HE | hematology | Sight Diagnostics , Ltd. | Sight OLO |
| K201301 | 10/2/2020 | 2020 | 510(k) | JOY | HE | hematology | Scopio Labs , Ltd. | X100 with Full Field Peripheral Blood Smear (PBS) |
| K200595 | 10/16/2020 | 2020 | 510(k) | JOY | HE | hematology | CellaVision AB | CellaVision DC-1, CellaVision DC-1 PPA |
| K202089 | 10/28/2020 | 2020 | 510(k) | POV | HE | hematology | Bonraybio Co., Ltd. | LensHooke X1 PRO Semen Quality Analyzer, LensHooke |
| DEN200080 | 9/21/2021 | 2021 | De Novo | QPN | PA | wsi_ai | Paige.Ai | Paige Prostate |
| P190022 | 12/7/2021 | 2021 | PMA | QRF | Immunology | molecular_ivd | Opko Health, Inc. | 4Kscore Test |
| K200828 | 3/22/2022 | 2022 | 510(k) | GKZ | HE | hematology | Athelas, Inc. | Athelas Home |
| K220013 | 5/3/2022 | 2022 | 510(k) | JOY | HE | hematology | Scopio Labs , Ltd. | X100HT with Slide Loader with Full Field Periphera |
| P210011 | 4/28/2023 | 2023 | PMA | PQP | Pathology | molecular_ivd | Tempus Labs, Inc. | xT CDx |
| K222148 | 8/16/2023 | 2023 | 510(k) | GKZ | HE | hematology | Pixcell Medical Technolog | HemoScreen Hematology Analyzer |
| K221309 | 9/19/2023 | 2023 | 510(k) | JOY | HE | hematology | Sigtuple Technologies Pvt | AI100 with Shonit |
| K230878 | 11/14/2023 | 2023 | 510(k) | GKZ | HE | hematology | Ad Astra Diagnostics, Inc | QScout Lab; QScout RLD |
| DEN210035 | 1/31/2024 | 2024 | De Novo | QYV | PA | cytology | Hologic, Inc. | Genius Digital Diagnostics System with the Geniu |
| K240636 | 5/2/2024 | 2024 | 510(k) | GKZ | HE | hematology | Pixcell Medical Technolog | HemoScreen Hematology Analyzer |
| P230009 | 7/26/2024 | 2024 | PMA | PHP | Medical Genetics | molecular_ivd | Guardant Health, Inc. | Shield |
| P230043 | 10/3/2024 | 2024 | PMA | PHP | Pathology | molecular_ivd | Exact Sciences Corporatio | Multi-Target Stool DNA (mtsDNA) Test, Cologuard Pl |
| K241232 | 1/24/2025 | 2025 | 510(k) | QPN | PA | wsi_ai | Ibex Medical Analytics , | Galen Second Read |
| K241717 | 2/28/2025 | 2025 | 510(k) | PSY | PA | wsi_infra | Shandon Diagnostics Limit | E1000 Dx Digital Pathology Solution |
| K242388 | 5/9/2025 | 2025 | 510(k) | POV | HE | hematology | Bonraybio Co., Ltd. | LensHooke X12 PRO Semen Analysis System |
| K242830 | 5/30/2025 | 2025 | 510(k) | POV | HE | hematology | Bonraybio Co., Ltd. | LensHooke X3 PRO Semen Quality Analyzer; LensHooke |
| K251371 | 6/25/2025 | 2025 | 510(k) | GKZ | HE | hematology | Sysmex America, Inc. | Sysmex XR-Series (XR-20) Automated Hematology Anal |
| K243144 | 6/27/2025 | 2025 | 510(k) | JOY | HE | hematology | Scopio Labs , Ltd. | X100/X100HT with Full Field Peripheral Blood Smear |
| DEN240068 | 7/31/2025 | 2025 | De Novo | SFH | PA | wsi_ai | Artera, Inc. | ArteraAI Prostate |
| P250005 | 8/15/2025 | 2025 | PMA | SFL | Medical Genetics | molecular_ivd | Biocartis Us, Inc. | Idylla CDx MSI Test |
| K250003 | 8/29/2025 | 2025 | 510(k) | PZM | PA | molecular_ivd | Geneseeq Technology, Inc. | GENESEEQPRIME NGS Tumor Profiling Assay (FFPE) (GS |
| K241868 | 9/19/2025 | 2025 | 510(k) | PZM | PA | molecular_ivd | Tempus AI, Inc. | xR IVD |
| K243348 | 2/6/2026 | 2026 | 510(k) | GKZ | HE | hematology | Athelas, Inc. | Athelas Home |
| K252228 | 4/9/2026 | 2026 | 510(k) | POV | HE | hematology | Checkcells, Inc. | Seaman Pro/Seaman |