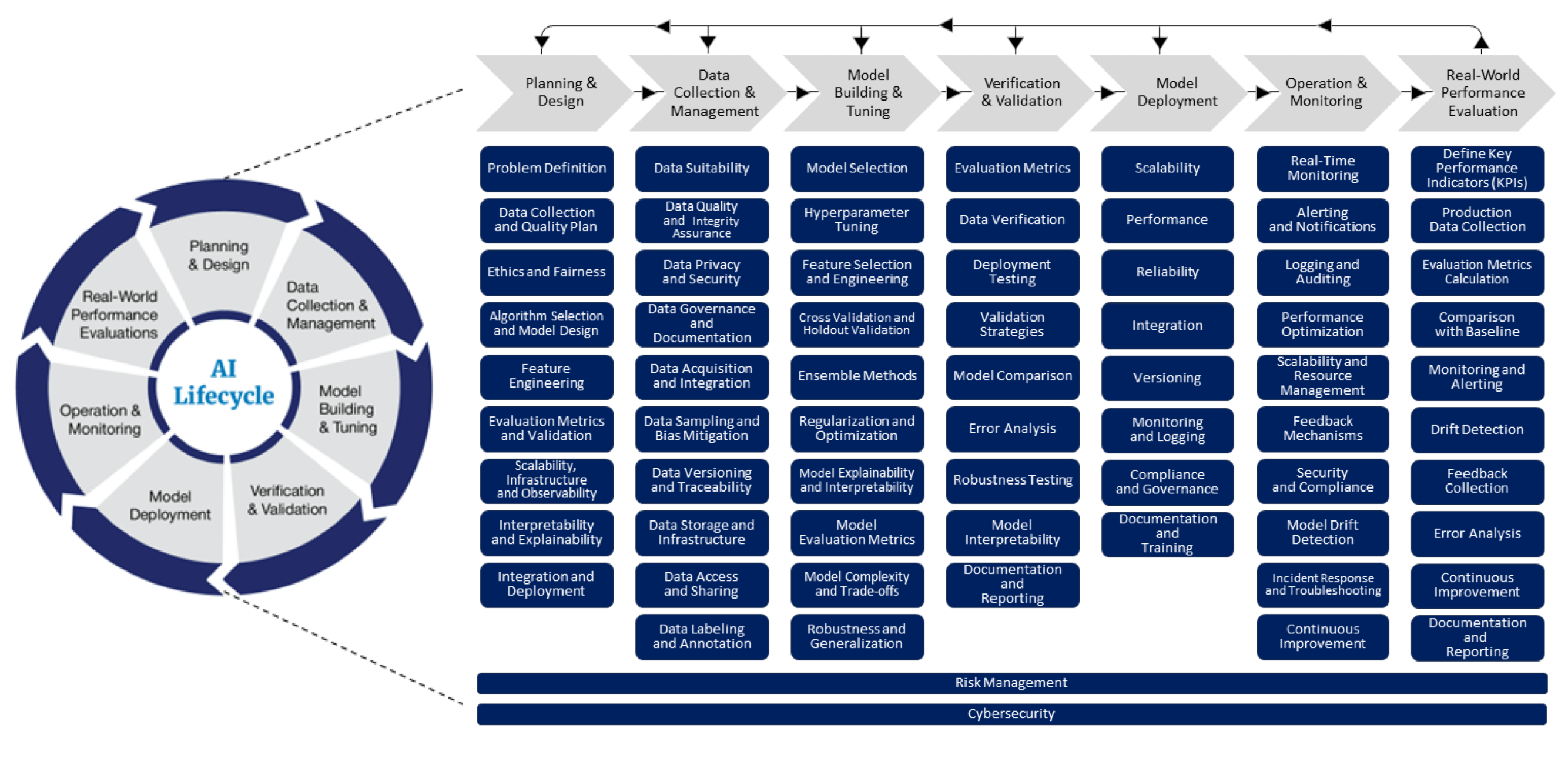
Developing medical-imaging AI applications has unique challenges. These challenges are difficult for startups to address with small in-house teams or generalist development firms. Here are some of the unique challenges companies in this space will need to overcome:
Challenge: AI Performance Validation
FDA requires clinical validation of most AI models. Inexperienced teams can lead to costly delays. Common problems include:
Insufficiently diverse training or testing data
Inappropriate data management or controls
Insufficiently rigorous reference standard
Inappropriate validation study design
To ensure your models get past FDA review, it is critical to have a team that understands FDA’s expectations around training data coverage, data controls and AI lifecycle processes.
FDA is placing an increasing focus on the AI development lifecycle
within their regulatory review.
How Innolitics Meets this Challenge
We have 11 engineers (including 3 PhDs) with deep image-processing expertise in traditional image processing and AI-based methods.
Our team of six FDA regulatory consultants have helped over 100 companies FDA clear medical device software. We have a deep understanding of the relevant FDA guidance and regulations.
What our Clients Say
During a presubmission meeting, the FDA acknowledged that Innolitics were “extremely advanced” in their understanding of the regulations. The FDA essentially concurred with all of Innolitics’ advice, demonstrating a deep understanding of the regulations, even in the face of ambiguous topics such as CADe vs non-CADe. They expertly, and politely, navigated the FDA call with our best interests in mind while fostering a collaborative exchange with the agency.
Dr. Ilya Pyatnitskiy, MD
Cofounder of AUMI AI
I worked with Innolitics to develop an application that analyzes MR images of our geometric distortion phantoms. It was a quite challenging image-processing project. In the end my company and Innolitics team were able to meet the challenge and built a quality web UI and a robust analysis pipeline. Innolitics seems like a company that is run by engineers who care about doing quality work.
Mircea Lazea
Technical Product Manager - Therapeutic Physics at Mirion Medical
The work conducted by Innolitics to complete a complex software port of a core module of our clinical product was outstanding. The clinical product must by cleared by the FDA, and as such, we needed the highest quality workmanship, and close collaboration with a dispersed team. Innolitics performed exceedingly well in gathering requirements, researching, developing and implementing a design, and providing test modules and documentation. Their team’s understanding of medical imaging analysis software, and FDA quality processes, was key to the success of the project.
Nick Schmansky
Co-Founder and CEO of CorticoMetrics
Innolitics has been invaluable in developing some components of our cloud-based analysis software for preclinical imaging. They expertly implemented an AI tool for the analysis of DICOM data and helped us with training and optimizing a prototype neural network. Their developers work independently, handling complex challenges with ease, and deliver top-quality results. It’s been a pleasure working with the dedicated and professional team at Innolitics, and we highly recommend their services.
Alex Klose, PhD
CTO and Co-Founder at InVivo Analytics
They were proactive, informative, and very well-versed in the regulatory challenges of AI in medical imaging… Unlike competitors, who sometimes take a blanket approach, Innolitics didn’t throw the baby out with the bathwater. They recognized the good work already done and built upon it… We reviewed other capable firms, but none showed the same depth of understanding of the rapidly evolving landscape of AI and regulatory requirements. Their proactive stance with the FDA gave us confidence that we were in the right hands, compared to competitors who seemed more reactive.
A practical guide for engineers new to medical imaging AI, highlighting common pitfalls—like coordinate-system errors and orientation mix-ups—and providing simple checks to avoid them.Ask ChatGPT
Setting acceptance criteria incorrectly (too high or too low) can delay your FDA submission by weeks or trigger rejection. This guide analyzed 784 AI/ML medical device clearances to provide concrete, data-driven thresholds. You’ll find specific starting points: AUC of 0.9, Sensitivity/Specificity of 0.8, Dice of 0.8, Kappa of 0.85, and Bland-Altman LOA within ±10% of mean physiological values. Learn which statistical methods FDA accepts most often, how to justify your criteria using special controls and predicates, and what to do when your device fails testing.
This article outlines the process of developing an AI/ML algorithm from scratch and getting it FDA cleared. It covers the four phases of the process (Explore, Develop, Validate, and Document) and discusses the costs, time, and data requirements involved. It also provides advice on regulatory strategy, data annotation, and algorithm prototyping. If you’re interested in developing a medical device involving AI/ML.
The article outlines a method for documenting AI/ML algorithms for FDA pre-submissions. It emphasizes clear algorithm descriptions to avoid delays. Key steps include visual runtime descriptions, dataset analysis, non-ML testing, annotation details, AI/ML test plans, and performance metrics. It stresses reducing annotation costs and leveraging existing data while advising comprehensive documentation for effective FDA evaluation.
Here we analyze over 200 FDA 510(k) and De Novo summaries to tease out best practices for AI/ML study designs and adjudication strategies specifically.
This article explains Bland–Altman analysis for evaluating agreement between two continuous measurement methods, especially in FDA-facing validation. It shows how bias and 95% limits of agreement reveal whether methods are interchangeable, why correlation and AUC can mislead, how to spot proportional bias, and includes practical interpretation tips, examples, and reusable Python code.
This article shows how to achieve an AI/ML SaMD 510(k) submission in ~3 months by running a compact, well-powered MRMC study while continuously accruing standalone evidence through washout. It clarifies roles, sizing, units, and endpoints, with device examples, flowcharts, and a checklist to prevent rework and delays.
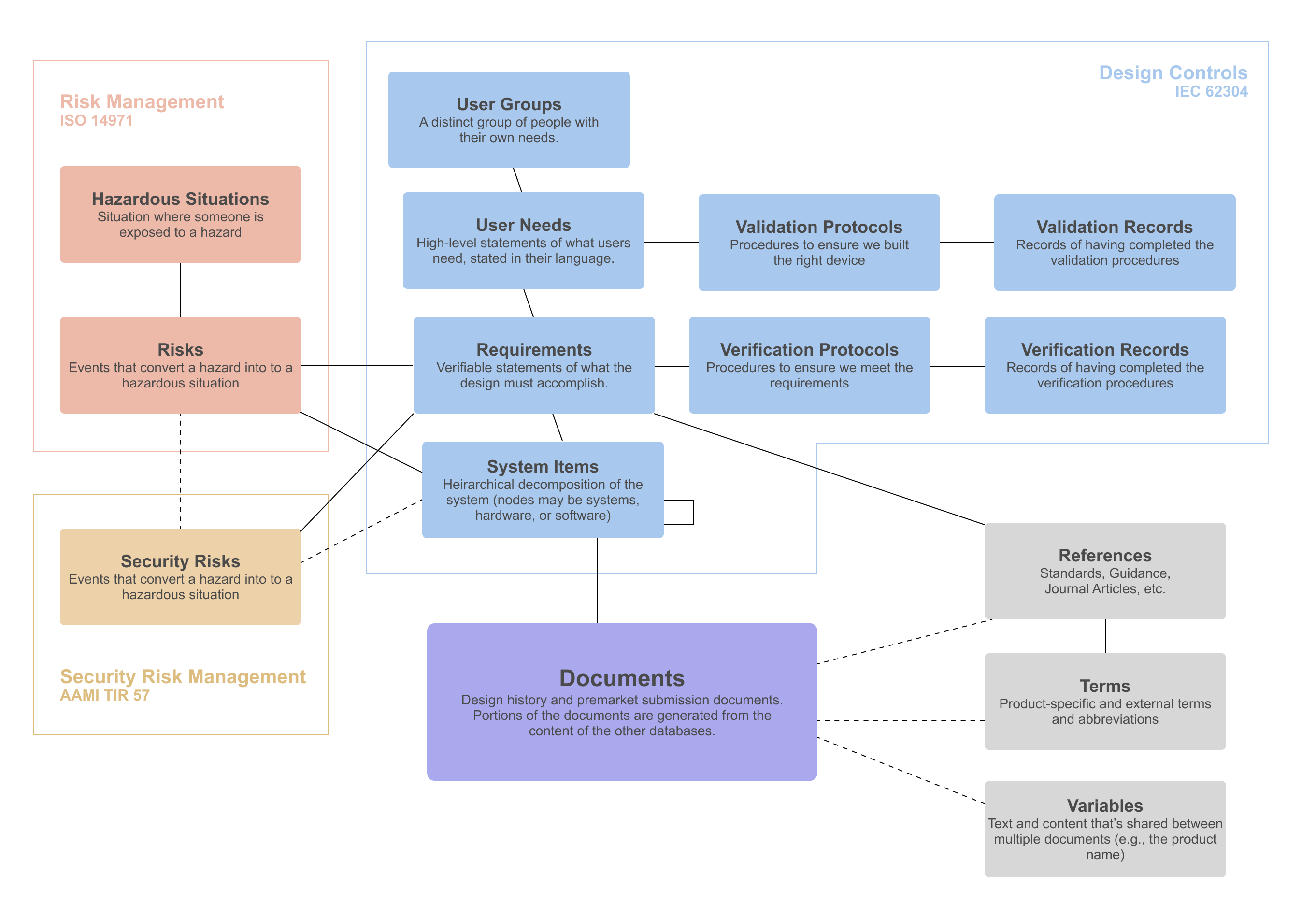
Challenge: Design Documentation for FDA Submissions
When you submit your 510(k) or De Novo application to FDA, you’ll need to provide comprehensive documentation of your product's development process, risk management activities, software architecture, and verification and validation testing.
It is not uncommon for companies to take 9 - 12 months just producing the documentation! This problem is so common that we offer our Fast 510(k) service to companies who are done with development but who didn’t produce the necessary design documentation. This service typically takes 3 months. For example, we’ve had to take over projects developed by over-seas teams who were unable to comply with this rigorous documentation.
It is much more efficient to pay more up front for a development team that can provide all of the necessary design documentation.
Diagram showing the various information that needs to be documented and
traced together during medical device AI development.
How Innolitics Meets this Challenge
Our Medtech OS platform is purpose built for medical device software submissions. Our engineers are trained on its use and it provides all of the documentation required for FDA submissions.
We’ve helped over 100 companies FDA clear their medical device software and have a thorough understanding of the relevant guidance and standards.
What our Clients Say
Thanks to Innolitics, we were able to complete the software documentation for our 510(k) in just 4 months. They flew onsite to absorb how our software worked and provided detailed guidance. Without Medtech OS, training, and support, I can easily imagine the process taking 12 months. Our engineering team was able to focus on feature development instead of being bogged down with documentation. We tried three different consultants before finding Innolitics. None of them could deliver. Innolitics took the reins and made it happen. I don’t see how we would have completed our 510(k) without them.
VP of Systems R&D
IVD Startup
I engaged Innolitics to develop a companion app for a COVID testing kit sold directly to users. The app’s purpose was to walk people at home through using the test kit. Innolitics was a pleasure to work with. They worked systematically, beginning with a planning phase to collect and clarify our user needs and requirements and ending with thorough V&V and documentation. I’m proud of the app we built together and would certainly work with them again.
Trudy Estridge, PhD
Senior Director Regulatory Affairs
Our FDA submission deadline was just two weeks away, and we had no software or cybersecurity documentation. We feared we would miss the deadline. Then our regulatory team introduced us to Innolitics. Their team swiftly validated our software and prepared the 15 necessary software and cybersecurity documents. They took a pragmatic approach that truly added value. This rapid timeline would have been impossible to meet without a team deeply knowledgeable in software, cybersecurity, AI/ML, and FDA regulations. In the end, we were able to submit on time! Thank you, Innolitics, for your Herculean efforts!
Andrea Cubitt
CEO of Dionysus Digital Health
7 weeks to submission and 6 months to FDA clearance. Innolitics delivered exactly what we needed. They took ownership of the entire DHF and 510(k) package—documentation, cybersecurity, risk management, the works. When we hit internal roadblocks, they found ways to keep us moving. When scope decisions needed to be made fast, they made the right calls. Fast, thorough, no surprises, and full service. That’s how regulatory consulting should work!
This checklist is meant to assist in the review of a 510(k) submission. It is a version of the checklist we use internally at Innolitics when we’re reviewing 510(k) submissions for our clients. The checklist applies to any SaMD 510(k) submissions made for medical devices.
This article dissects a real AI/ML SaMD 510(k) submission (Body Check CT Cardiomegaly, K232613) with downloadable examples. After wasting a year over-engineering my first submission, I’ll share practical insights to prevent this from happening to you!
With predetermined change control plans (PCCPs), FDA has given manufacturers a great new regulatory tool. In this article, we discuss PCCP best practices and questions based on our experiences with FDA and our thorough research of the FDA databases.
This article outlines Innolitics’ process for developing machine learning algorithms for FDA-cleared medical devices. It covers documentation requirements, common pitfalls (overfitting, data leakage, augmentation errors), and a systematic workflow using four key reports: Input Verification, Data Augmentation QA, Model Performance, and Model Comparison—ensuring safe, effective, and compliant AI development.
Integrating within the clinical workflow is critical for success. Doing so requires an understanding of DICOM, HL7, and FHIR. It also requires a real-world understanding of hospital IT environments. Although acceptance of cloud-based deployments is growing, many institutions still require on-site deployments.
Furthermore, performance is critical. Radiologists are busy, highly paid professionals with low tolerance for waiting. They will not wait for large images to load. Solving these performance issues is challenging given the size and quantity of image sets (MRIs and CTs).
How Innolitics Meets this Challenge
We have over 5 years of experience deploying and maintaining AI-enabled radiology software.
We are DICOM experts (e.g., our DICOM Standard Browser is used by over 10,000 people each month).
What our Clients Say
We engaged Innolitics as a software partner for our AI/ML-enabled radiology application. Their knowledge of AI/ML deployments, DICOM, and FDA regulations added a lot of value on a tight pre-seed budget. We’d definitely recommend them for their niche expertise in medical-device AI/ML.
Dr. Raj Shah, MD, MBA
Co-Founder of GuidewireRX
Innolitics’s engineers seamlessly integrate with our existing team. They’ve consistently produced high quality work while helping us solve technical problems in the medical imaging space. In particular, they spearheaded development of a large module that extends our existing product suite. This module has since been cleared by the FDA and is live at hospitals around the world. Their work on the module involved software development, researching various image processing algorithms, analysis of these algorithms against real-world data, and writing detailed technical documentation of the system that justified its approach to the FDA.
Nathan Childress, PhD, DABR
Associated VP at Varian Medical Systems
I have really enjoyed working with the Innolitics team. We had originally enquired about the Innolitics DICOM conformance statement consulting but expanded them to augment the data engineering team. They helped build out a complex ETL pipeline for AI/ML and helped build out accompanying SOPs for regulatory compliance. They treat our product as if it was their own and integrate seamlessly with our in-house engineering team. Their breadth of knowledge in technical and regulatory matters has helped our team meet deadlines.
Dr. Heling Zhou, PhD
Director of Data Engineering at Enlitic
We commissioned Innolitics to build a dashboard that integrated with our radiation-oncology EMR. Their team’s clinical experience greatly simplified communication and their perceptive questions and suggestions helped us arrive at a better set of requirements for the project. The final result was clean, easy for our doctors to use, and has already improved the efficiency of our workflow.
Jonas Fontenot, PhD
President and CEO of Mary Bird Perkins Cancer Center
A practical guide for integrating software with VistA, the VA’s PACS. This guide is based on our experience working through the process with our client.
Overview of SMART on FHIR and when it’s a good approach to integrating your software into the clinical workflow. It includes a few sequence diagrams and discusses when its an appropriate fit for your device.
The FDA says developing your design inputs is “the single most important design control activity,” yet writing good design inputs is difficult. This article presents Innolitics’ answers questions our clients frequently ask us about design inputs and analyzes a number of poorly written example requirements.
An in-depth guide to navigating the use of off-the-shelf software (OTS) in the medical device industry. OTS software is general purpose software you didn’t develop yourself that you use in your device, e.g., open-source packages, cloud services, and operating systems. The article starts with the basics and then delves into more detailed issues, including strategies for documenting OTS software for the FDA.
With an increase in malware-based attacks on hospitals and FDA’s new statutory authority to regulate medical device cybersecurity, vendors must focus increased resources on secure product development.
How Innolitics Meets this Challenge
We’ve helped dozens of companies resolve FDA cybersecurity deficiencies and have a keen understanding of FDA’s guidance and requirements.
We can handle all aspects of cybersecurity, including threat modeling, security risk management, cybersecurity controls, SBOM generation, and cybersecurity testing.
What our Clients Say
Innolitics is an incredible partner and consistently surpasses our expectations. They have an extremely agile team, adapting to our needs across back-end and front-end tasks seamlessly. When we needed support around ISO 62304 compliance for FDA requirements, they jumped right in and provided us compliant documentation. They also assisted us as we developed a regulatory strategy around FDA Cybersecurity and HIPAA Compliance. The Innolitics team is efficient, fair, and highly ethical. They are an absolute pleasure to work with.
Ryan Shelton, PhD
CEO and Co-Founder of PhotoniCare
With updated cybersecurity requirements rolled out during our recent FDA submission, we found ourselves looking for an experienced partner to help us navigate this new environment. We reached out to Innolitics, and they were able to quickly assess our device, develop a strategy, and meet with FDA to find a path forward. Their strategic involvement guided us toward an approach that satisfied the FDA and saved us significant upfront and on-going effort. Innolitics’ combination of software and FDA regulatory expertise was invaluable.
Eric Runde
COO at Indica Labs
We really enjoyed working with Innolitics in reviewing our cybersecurity related documentation and compiling a response to the FDA. We were particularly impressed with their knowledge and expertise in the field, and quick turn-around times to our queries. We look forward to working with Innolitics on future projects.
How do you avoid slowing down your FDA marketing submission (510(k), De Novo, or PMA) with cybersecurity problems? This article reviews 14 common FDA cybersecurity deficiencies based on our recent submissions.
This article provides an in-depth exploration of medical device cybersecurity requirements, including best practices and FAQs. It also includes examples and resources for those looking to implement or improve their own threat modeling processes.
This is a redline between the 2022 Draft cybersecurity guidance and the final 2023 cybersecurity guidance. It should be useful to medical-device regulatory professionals who want to see, in detail, what has changed since the draft guidance.
How safe is DICOM in the wild? As you develop your medical imaging device or service, prepare for the standards and regulations protecting hospital networks from ransomware.
We can be your full-service software partner for a new medical imaging AI product.
We will work with your team to design, develop, validate, FDA clear, and maintain the software for your new medical-device AI product.
It is common for startups to have a couple of founding engineers who will manage the core AI model. In these cases, we can coordinate with your internal team while we develop the surrounding UI, deployment scaffolding, and cybersecurity requirements for the project.
If you don’t have a regulatory consultant, we can also be your regulatory partner. Read more about our regulatory services here.
We can work with your existing engineers or data-scientists to complete software development projects with a defined scope. Typical projects include:
Training novel AI models
Local and cloud deployments
Medical image viewer development
EMR, PACS and worklist integrations
HIPAA and FDA cybersecurity support
Validating and tuning research-grade image-processing models
This engagement model works well when the scope of the project is understood. We will work with you to define a scope, cost-estimate, and timeline estimate for the project.
We provide experienced medical-device software engineers to augment your in-house team.
Our engineers are trained on the FDA regulations and guidance and can work within your QMS.
We provide engineers at either half-time or full-time and at varying levels of experience. We can also increase and decrease staffing levels as needed.
This engagement model is appropriate if you have poorly scoped work or need ongoing support. Often, after completing a project-based engagement, our clients request to retain the engineers they work with in an on-going basis.
Medical Imaging Libraries: pydicom, ITK, VTK, SimpleITK, DCMTK
Containerization and Orchestration: Docker, Kubernetes
Some of the technologies we use when developing medical imaging AI for
our clients.
FAQ
Can you help us collect and annotate data for our model?
Yes, since we’ve been working in the radiology space for over a
decade, we have a broad network of data providers and clinicians. In
most cases, we can help procure data and clinical specialists to help
annotate the data.
Do you work on non-imaging AI applications?
Yes! Although our deepest expertise is in the imaging domain, we also
have experience with IVDs and other signal-processing applications.
Let’s talk and we can explore if we’re a good fit for your needs.
How much does your typical project cost?
End-to-end projects can vary quite a bit depending on several
factors:
Whether the core algorithms or models have been proven
The size and complexity of the user interface (if any)
The number of platforms that are needed
The risk-classification of the device (higher-risk devices require
more documentation and testing)
We do not work on a fixed-price basis for large projects, but in some
cases we may begin projects with a fixed-price scoping phase or a small
fixed-price initial engagement.
For our project-based engagements and end-to-end engagements we
provide detailed cost and timeline estimates. We refine and periodically
update these estimates throughout the project.
Can you provide examples of past projects similar to our product?
Yes! Please review our
case studies for a sampling of our past projects. If you don’t see
anything relevant, please reach out as only a small number of our
projects have case studies.
How do you handle project management and communication during the development process?
For staff-augmentation projects we assume you will be managing our
resources along with the rest of your team.
For our project-based and end-to-end engagements we provide project
management support. Here are a few of the highlights:
We set up preferred communication channels during kickoff (often
Slack or Microsoft Teams)
We follow an agile process with one- or two-week sprints
During these meetings we demo our work and plan the next sprint
We provide periodic budget and timeline estimate updates
What experience does your team have with FDA regulatory submissions for medical devices?
We have deep experience with FDA regulatory submissions. We have six
full-time regulatory consultants on our team. Collectively we have
cleared over forty-five 510(k) submissions and several De Novos. Our
engineers are also trained on writing the software and cybersecurity
design history documentation required for FDA submissions.
In short, we are among the most experienced FDA regulatory experts in
the world when it comes to medical device AI software.
Does your team do firmware development?
Several engineers on our team have experience with firmware
development, however, we typically are not the best fit for
firmware-heavy projects. We excel on Software in a Medical Device (SiMD)
projects that involve a lot of application-level coding, such as web,
mobile, or desktop UIs, cloud deployments, image processing, etc.
Can your team help design our user interface?
Yes. Typically we will develop an initial set of UI Mockups in Figma
and will then have a Medical Device UI/UX designers polish and further
refine the slides.
What if your engineers don’t know a core technology that we use?
Good engineers can usually become proficient using new databases,
frameworks, or even programming languages pretty quickly. Tools like
GitHub Co-Pilot and ChatGPT have made it learning new technologies even
easier.
If this is a major concern, we may cover the cost of training
engineers on new technologies.
Can your engineers work within our QMS?
Yes! We can work within your QMS or within ours. We are happy to sign
a quality agreement to coordinate quality activities. We can also train
our engineers within your QMS. This often makes sense for Staff
Augmentation engagements.
Can you help us set up a Quality Management System (QMS)?
Yes. We can get you set up on our Medtech OS platform. This is the
platform we use for our QMS. You can read more about
Medtech OS here.
Can you run our QMS?
Sorry, although we’re often asked to do this, we do not currently
help companies run their QMS, however, we can provide guidance and
support. We do have several partners whom we can recommend who can help
you run your QMS.
An in-depth guide to navigating the use of off-the-shelf software (OTS) in the medical device industry. OTS software is general purpose software you didn’t develop yourself that you use in your device, e.g., open-source packages, cloud services, and operating systems. The article starts with the basics and then delves into more detailed issues, including strategies for documenting OTS software for the FDA.
The FDA says developing your design inputs is “the single most important design control activity,” yet writing good design inputs is difficult. This article presents Innolitics’ answers questions our clients frequently ask us about design inputs and analyzes a number of poorly written example requirements.
Are you losing customers because your software doesn’t integrate with their clinical workflow? This article provides regulatory and software tips for completing medical-device software integrations, helping you sell to more customers.
Medical-device software release frequency is a common question, particularly from software engineers familiar with agile development. The answer hinges on two key factors: whether the changes require regulatory submissions and the ability to produce all necessary design change documentation quickly.