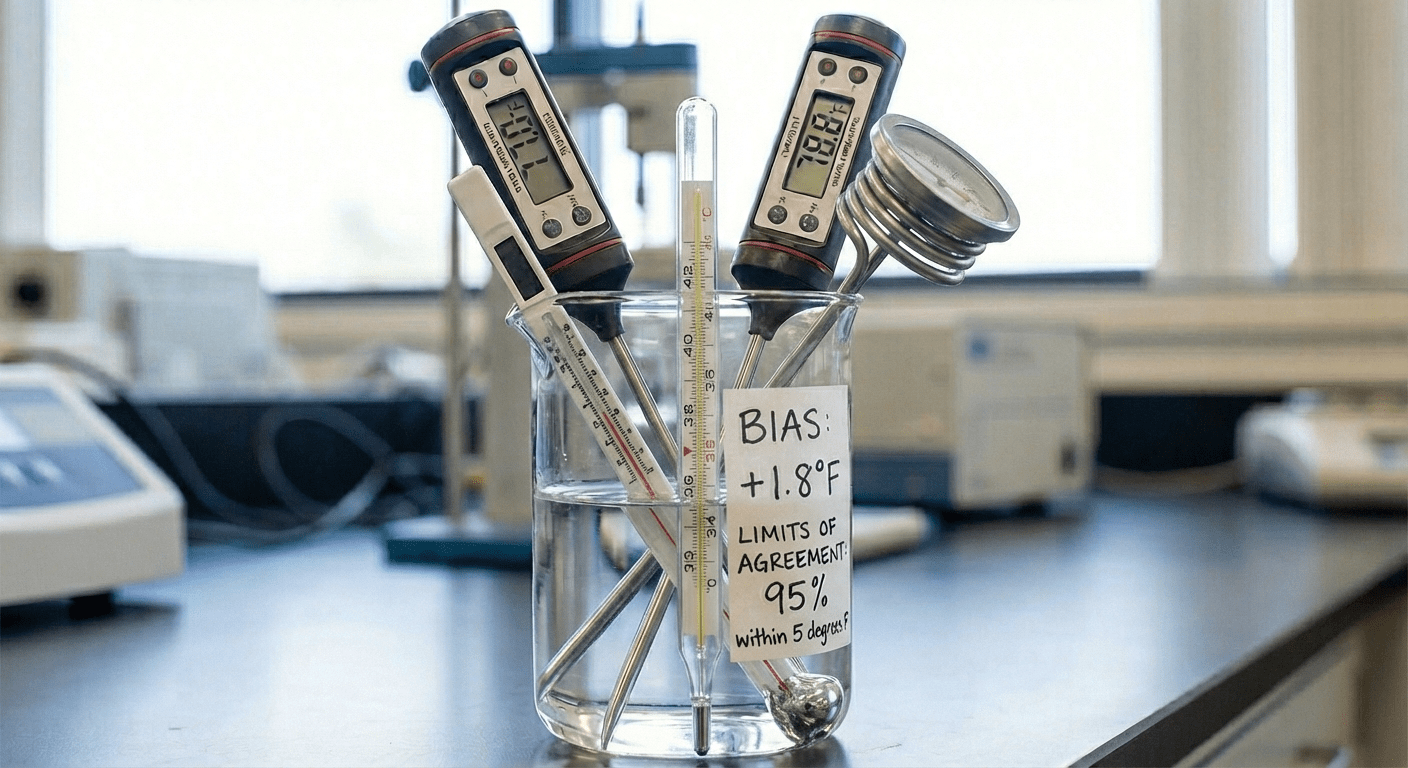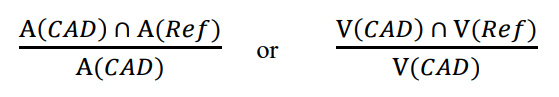FeaturedArticleAI/ML
How TEMPO and ACCESS Could Support Wearable Diagnostic AI
Most diagnostic AI teams clear FDA, then stall on reimbursement. FDA's TEMPO pilot and CMS's ACCESS model take on evidence and ...
 J. David Giese
J. David Giese
Medical device software articles, videos, and guidance from Innolitics.
FeaturedArticleAI/ML
Most diagnostic AI teams clear FDA, then stall on reimbursement. FDA's TEMPO pilot and CMS's ACCESS model take on evidence and ...
J. David Giese
FeaturedArticleAI/ML
Can a frontier AI model replace your FDA regulatory consultant? We built a private 1,200-question benchmark, an opinionated dis...
 Yujan Shrestha
Yujan Shrestha
FeaturedArticleAI/ML
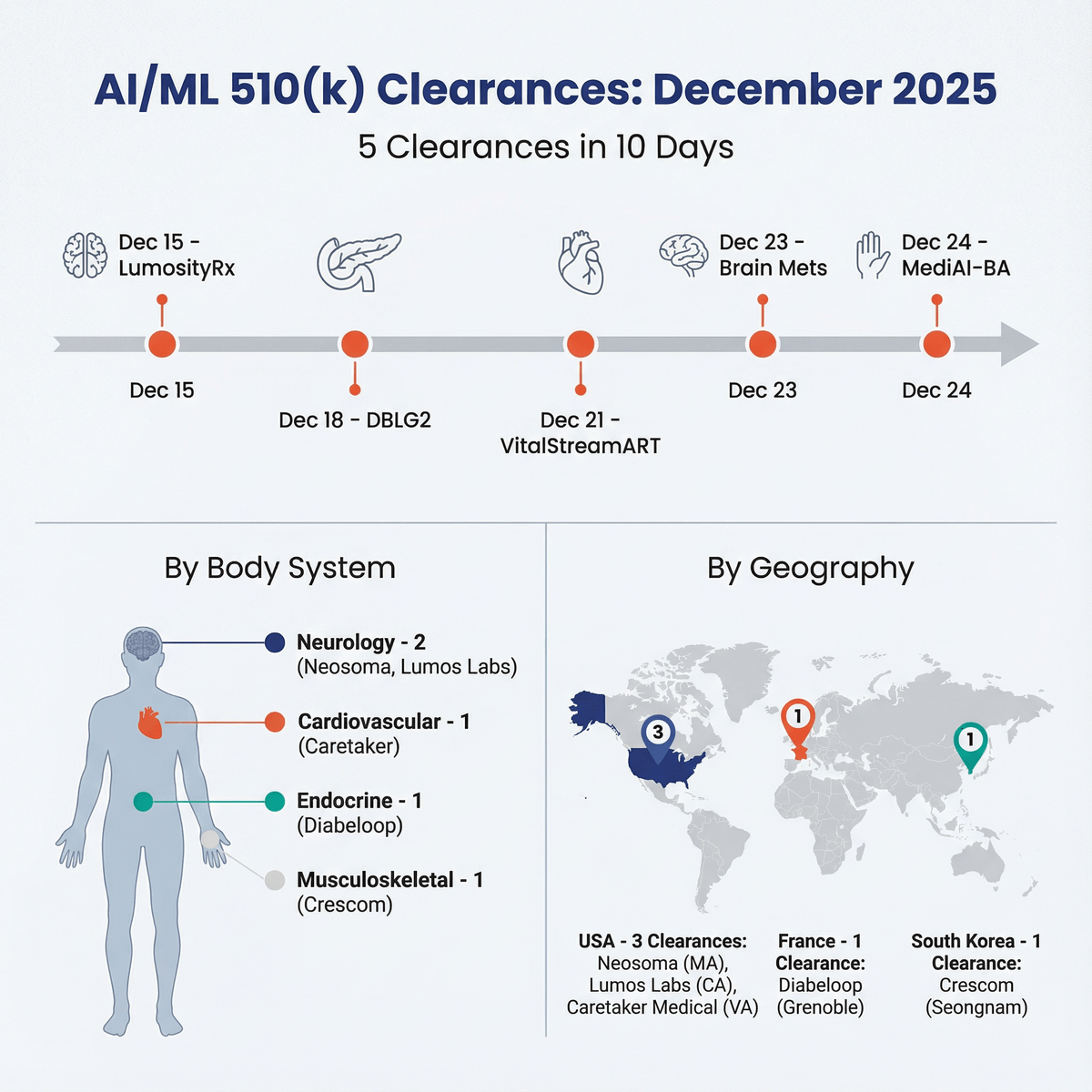
FDA authorized 86 AI/ML devices in Q2 2026, but the sharper signal came after first clearance: 12 Special 510(k)s, 12 PCCPs, tw...
Yujan Shrestha
FeaturedArticleAI/ML
FDA's draft MDUFA VI commitment letter (FY2028–2032) restructures how sponsors interact with the agency. Seven changes that wil...
Yujan Shrestha
FeaturedArticleAI/ML
UpDoc is now the clearest public example of an FDA-cleared LLM-enabled medical device. Here's what FDA actually cleared, why th...
Yujan Shrestha
FeaturedArticleAI/ML
For AI SaMD founders deciding how much to raise and how long the first FDA 510(k) will take. A 37-company benchmark: $16.0M mar...
Yujan Shrestha
FeaturedArticleAI/ML
Deep dive into four FDA De Novo and 510(k) submissions for AI-powered whole slide image analysis in pathology: Paige Prostate (...
 Maddie Tran & Yujan Shrestha
Maddie Tran & Yujan Shrestha
FeaturedArticleAI/ML
For founders, CEOs, and CFOs at AI/ML medical device companies trying to decide how much to spend on a 510(k). A meta-analysis ...
Yujan Shrestha
FeaturedArticleAI/ML
FDA cleared 24 AI/ML SaMD applications in March 2026 — one every 31 hours. A breakdown of the patterns that matter for sponsors...
Yujan Shrestha
FeaturedArticleAI/ML
FDA's glossary defines foundation models as AI models trained on large, typically unlabeled datasets that are adaptable across ...
Yujan Shrestha
FeaturedArticleAI/ML
Fifty-one FDA-authorized pathology AI devices through April 2026, only seven of which are whole-slide imaging algorithms. This ...
Yujan Shrestha
FeaturedArticleAI/ML
A deep dive into the 97 cleared cardiovascular AI/ML SaMD devices, the four archetypes that organize them, and what they reveal...
Yujan Shrestha
FeaturedArticleAI/ML
Why digital health companies keep dying before and after FDA authorization, the evidence-reimbursement trap strangling the sect...
Yujan Shrestha
FeaturedArticleAI/ML
AI-assisted medical device development is FDA-compliant when done right. The obstacle isn’t regulation. It’s the messy codebase...
Yujan Shrestha
FeaturedArticleAI/ML
Comprehensive analysis of 2025 cardiology AI/ML clearances, market trends, capital events, and regulatory insights.
Yujan Shrestha
FeaturedArticleRegulatory
Side by side differences and executive takeaways from the January 2026 updates to the FDA CDS and General Wellness updates
Yujan Shrestha
FeaturedArticleAI/ML
44 clearances. 18 in 2025 alone. The dental AI market has reached its tipping point. This report dissects the Pearl/Overjet duo...
Yujan Shrestha
FeaturedArticleAI/ML
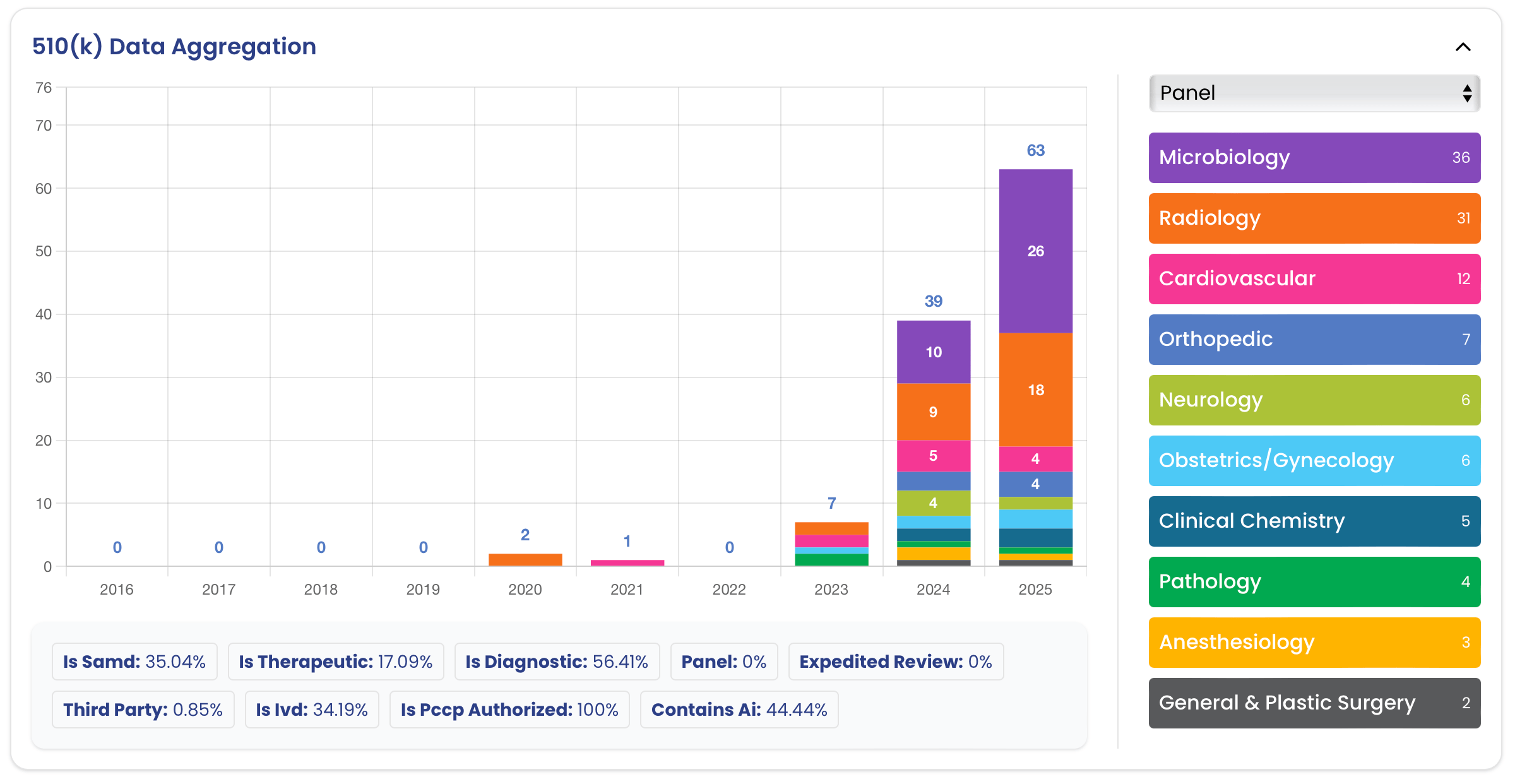
A data-driven look at 2025’s AI/ML 510(k) clearances: volume, review times, SaMD share, leading specialties, top manufacturers,...
Yujan Shrestha
FeaturedArticleAI/ML
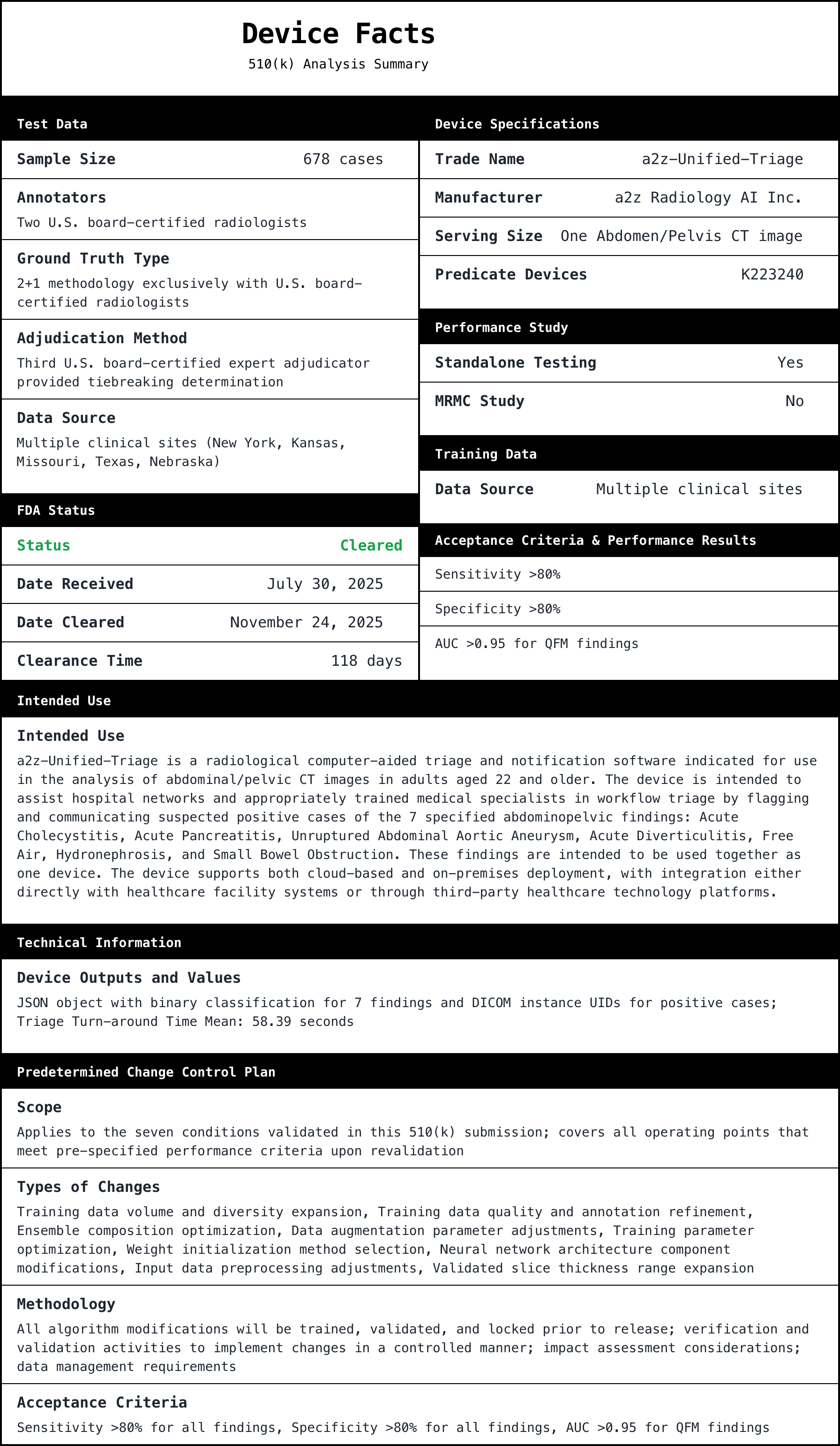
Deep dive into FDA 510(k) K252366, a2z‑Unified‑Triage, a radiology triage and notification device. Explains why the triage indi...
Yujan Shrestha
FeaturedArticleAI/ML
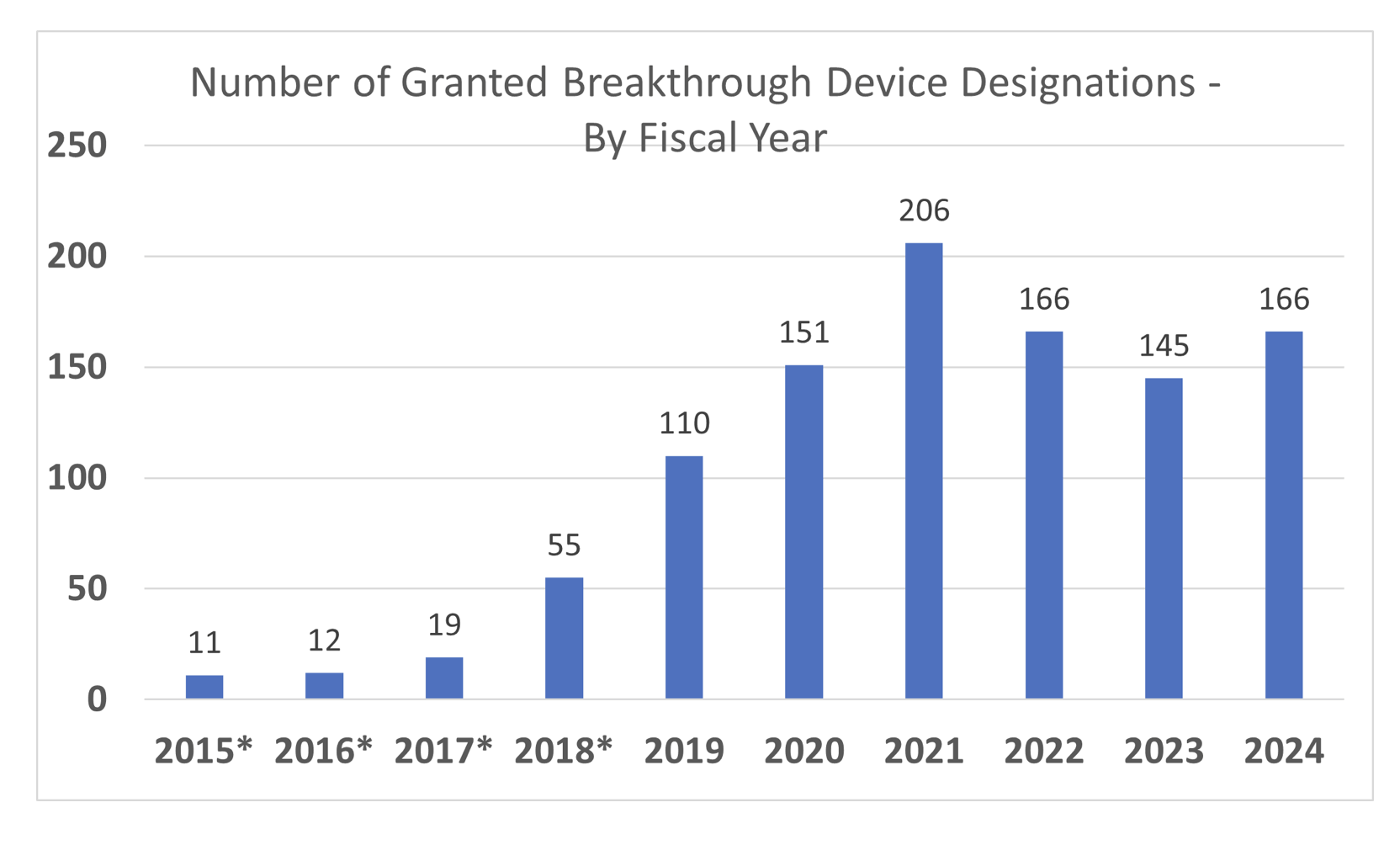
Breakthrough Device Designation (BDD) is a free, fast FDA program that signals serious unmet need and potential meaningful impr...
Yujan Shrestha & George Hattub
FeaturedArticleRegulatory
A Pre-Sub is a mechanism for requesting formal written feedback from the FDA, and (optionally) a one-hour meeting. Pre-subs are...
J. David Giese
FeaturedArticleAI/ML
Setting acceptance criteria incorrectly (too high or too low) can delay your FDA submission by weeks or trigger rejection. This...
Yujan Shrestha
FeaturedArticleAI/ML
This article shows how to achieve an AI/ML SaMD 510(k) submission in ~3 months by running a compact, well-powered MRMC study wh...
Yujan Shrestha
FeaturedArticleAI/ML
Here we analyze over 200 FDA 510(k) and De Novo summaries to tease out best practices for AI/ML study designs and adjudication ...
Yujan Shrestha
FeaturedArticleAI/ML
A practical guide for engineers new to medical imaging AI, highlighting common pitfalls—like coordinate-system errors and orien...
Yujan Shrestha
FeaturedArticleRegulatory
The FDA says developing your design inputs is “the single most important design control activity,” yet writing good design inpu...
J. David Giese
FeaturedArticleRegulatory
A concise guide to the FDA’s De Novo pathway for novel, moderate-risk medical devices that lack a suitable predicate. It includ...
J. David Giese
FeaturedArticleCybersecurity
This article provides an in-depth exploration of medical device cybersecurity requirements, including best practices and FAQs. ...
J. David Giese &
FeaturedArticleCybersecurity
Practical suggestions and tips for authoring SBOMs for medical devices and for using them to monitor for cybersecurity vulnerab...
J. David Giese
FeaturedArticleAI/ML
Ideas on how to get foundation models and generative AI FDA cleared.
Yujan Shrestha
FeaturedArticleAI/ML
Recent advancements in radiology have led to the development of foundation models integrating vision and language capabilities,...
Yujan Shrestha
FeaturedArticleRegulatory
A comprehensive FAQ for people who are interested in creating a new medical device software to learn the basics about the US re...
J. David Giese
ArticleAI/ML
With predetermined change control plans (PCCPs), FDA has given manufacturers a great new regulatory tool. In this article, we d...
J. David Giese
ArticleAI/ML
FDA publicly rejected Nihon Kohden's letter-to-file decision, so we pulled every warning letter FDA has published — 5,712 of th...
Yujan Shrestha
ArticleAI/ML
CMS and CDC are asking whether AI-assisted postanalytic interpretation, digital pathology, cloud analytics, and data-only medic...
Yujan Shrestha
ArticleRegulatory
A method for studying FDA guidance documents so you can give actionable, correct, and instant advice on a live call: read compl...
Yujan Shrestha
ArticleAI/ML
Learn how to write an FDA-ready AI model description covering inputs, outputs, architecture, training, selection, thresholds, p...
Yujan Shrestha
ArticleSoftware
Notes on medtech funding, covering accelerators and funding sources.
J. David Giese
ArticleAI/ML
27 AI/ML medical devices authorized in April 2026. Radiology's share of clearances dropped from 79% to 56%, QIH remains the reg...
Yujan Shrestha
ArticleAI/ML
TytoCare's De Novo cracked open ENT, a specialty with just five prior AI/ML clearances, and dropped a 510(k) on-ramp for everyo...
Yujan Shrestha
ArticleAI/ML
From solo clinicians prototyping algorithms to enterprise teams shipping production software, tools like Cursor, Claude Code, C...
Yujan Shrestha
ArticleNews
A reflection on how Innolitics revisits its mission and values at the annual retreat—and why that exercise sharpens judgment, s...
J. David Giese
ArticleCybersecurity
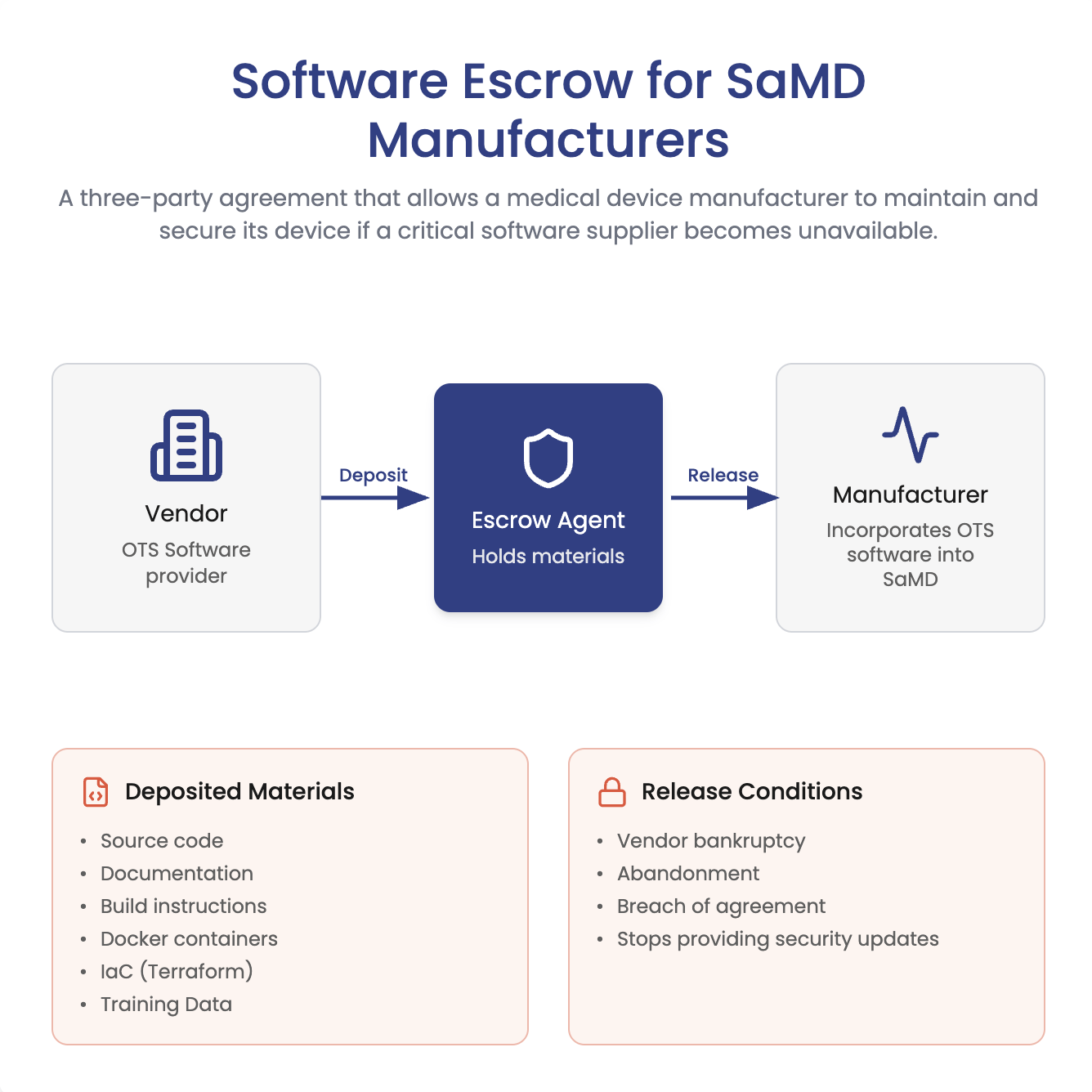
Does FDA require software escrow for SaMD? Not necessarily. Learn when escrow helps manage closed-source vendor risk, plus what...
J. David Giese
ArticleAI/ML
The U.S. Senate HELP Committee confirmed what we've been publishing for three years. An analysis of the regulatory signal, what...
Yujan Shrestha
ArticleAI/ML
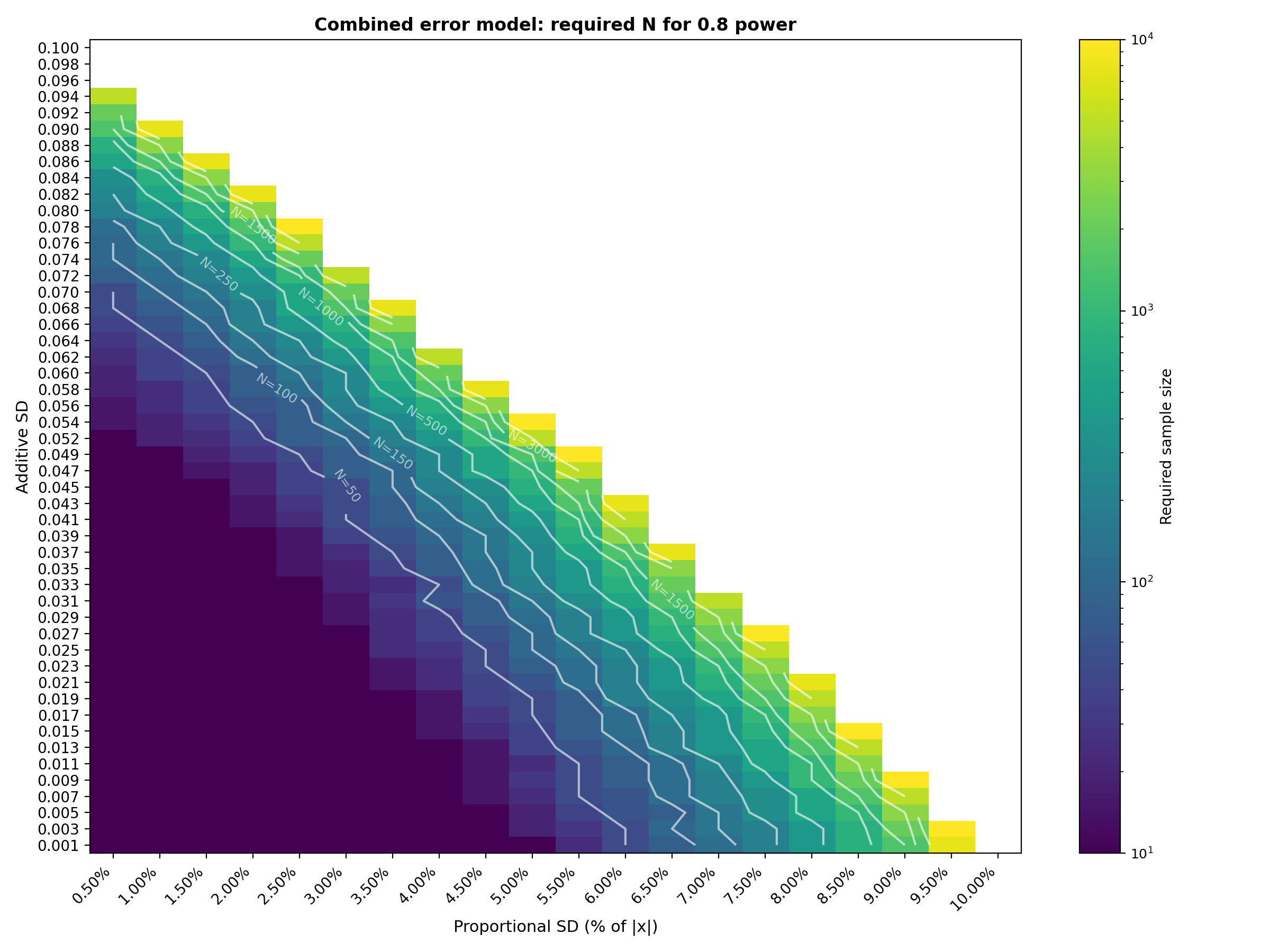
A step-by-step guide to using Monte Carlo simulation for estimating the sample size needed to pass Bland-Altman Limits of Agree...
Yujan Shrestha & Nishant Dahal
GuidanceCybersecurity
The 2026 FDA guidance on medical device cybersecurity, updated to align cybersecurity expectations with the Quality Management ...
GuidanceAI/ML
This FDA guidance clarifies which clinical decision support (CDS) software functions are excluded from the definition of a medi...
 Valeriia Zakrasniana
Valeriia Zakrasniana
ArticleAI/ML
Yujans commentary on the new Non-invasive blood pressure measuring device FDA guidance document in response to WHOOP and others.
Yujan Shrestha
ArticleAI/ML
A deep dive into K232613 (CT Cardiomegaly)
Yujan Shrestha
ArticleAI/ML
6 FDA clearances in 10 days. The AI pace is accelerating. Coverage includes neuro, dental, diabetes, and ADHD. Key wins: Nobel ...
Yujan Shrestha
Article
Diabeloop’s DBLG2 clearance proves high-risk SaMD viability, demonstrating how decoupling software from hardware minimizes regu...
Yujan Shrestha
ArticleRegulatory
Are your medical device timelines slipping despite a smart team and lots of effort? This article explains Innolitics’ practical...
J. David Giese
ArticleAI/ML
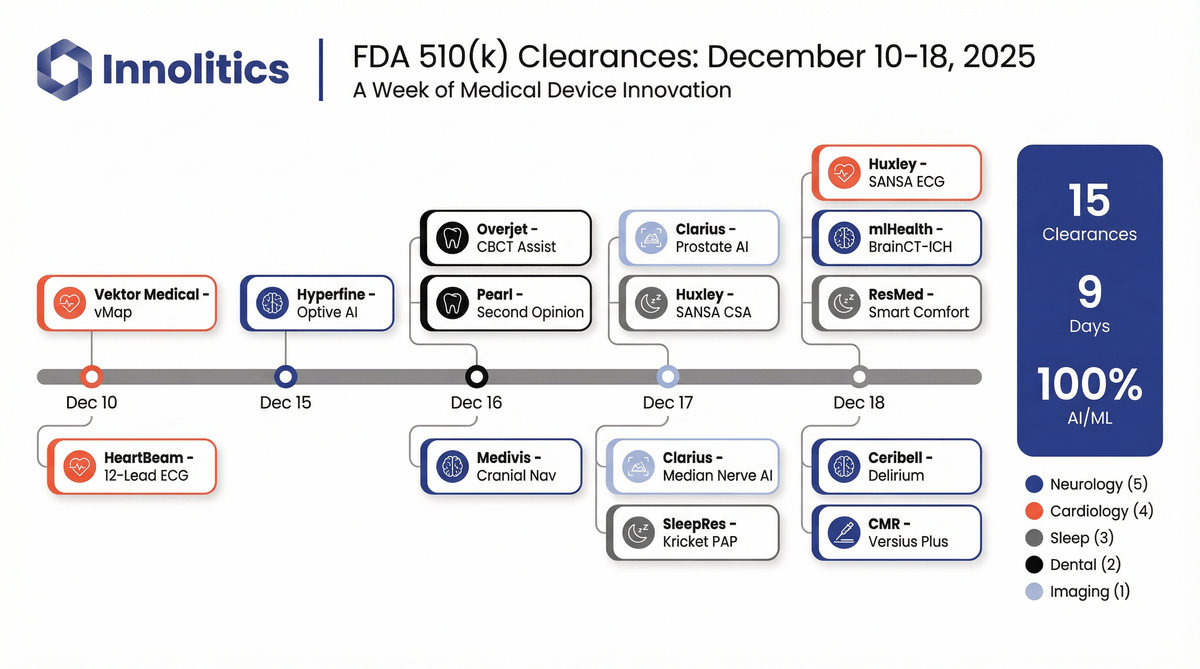
Fifteen AI/ML software 510(k) clearances landed in nine days in mid-December 2025. Here’s what got cleared, where it fits clini...
Yujan Shrestha
ArticleAI/ML
Analysis of K252086 (DTX Studio Assist), a strategic 510(k) clearance for a 'headless' SDK. This submission minimizes regulator...
Yujan Shrestha
GuidanceRegulatory
Learn FDA’s updated expectations for using real‑world data to generate real‑world evidence in medical-device submissions—includ...
Valeriia Zakrasniana
ArticleAI/ML
A client called out the “consultant thing”: hiding behind “ideally” instead of recommending a real plan. This piece explains wh...
Yujan Shrestha
ArticleAI/ML
This article explains Bland–Altman analysis for evaluating agreement between two continuous measurement methods, especially in ...
Yujan Shrestha
Article
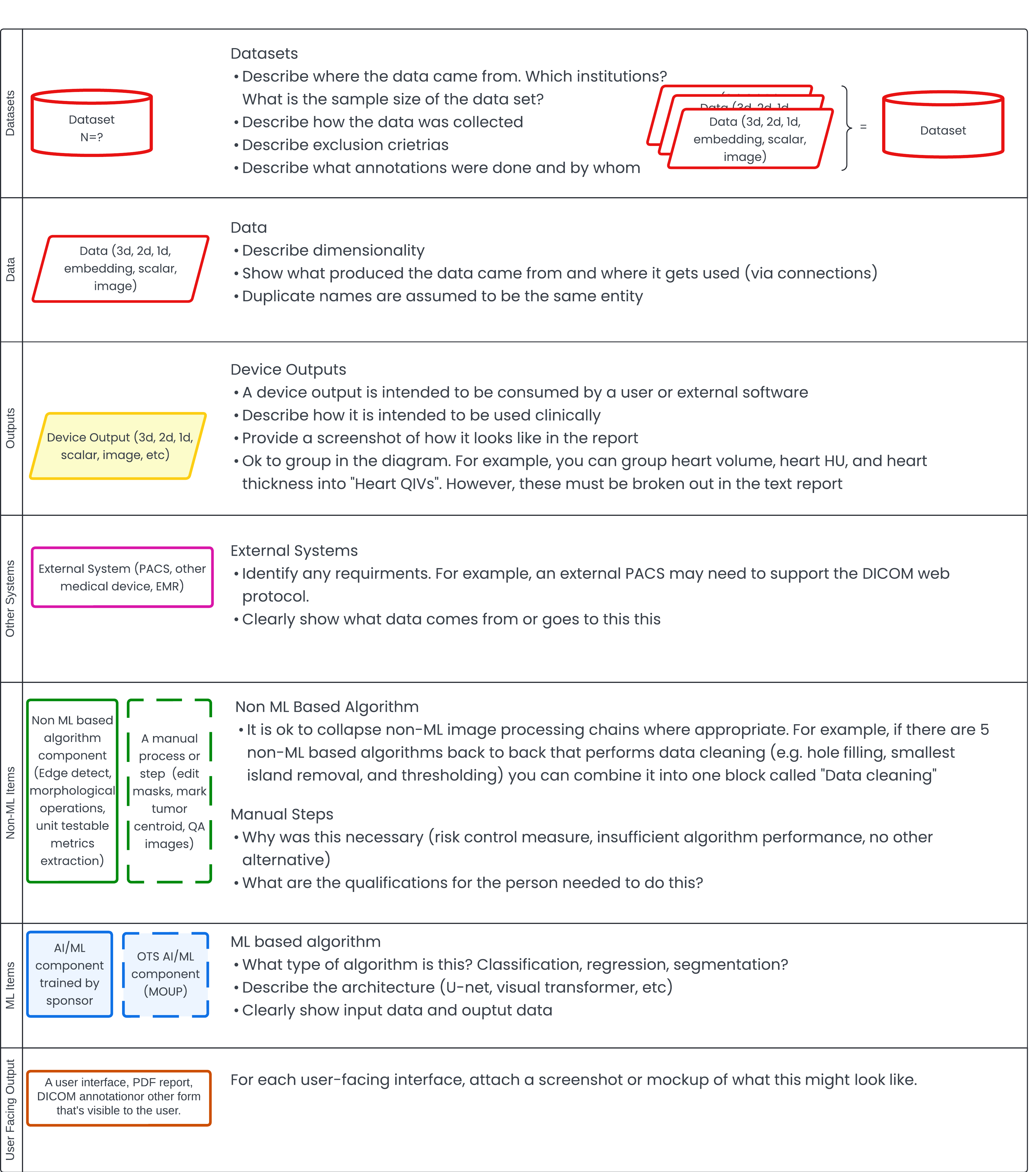
Practical tips on how to structure your SaMD architecture for FDA review without mirroring your codebase. Explains what the FDA...
 Meri Martinez & Yujan Shrestha
Meri Martinez & Yujan Shrestha
Article
A “quick” reference guide grouping the example 510(k) clearances by their product code/regulation, summarizing required testing...
Meri Martinez
ArticleAI/ML
Most first-time teams burn over a year overbuilding documentation FDA never asks for. Here is a section-by-section breakdown of...
Yujan Shrestha
ArticleRegulatory
An FAQ regarding the FDA’s new ACNU Rule.
J. David Giese
ArticleRegulatory
The “Unresolved Anomalies” document is required for the FDA’s pre-market submissions. This article includes best practices, FAQ...
J. David Giese
ArticleRegulatory
Wondering whether FDA’s Breakthrough Device Designation or STeP program is worth the effort? This pocket guide unpacks the prog...
J. David Giese
ArticleAI/ML
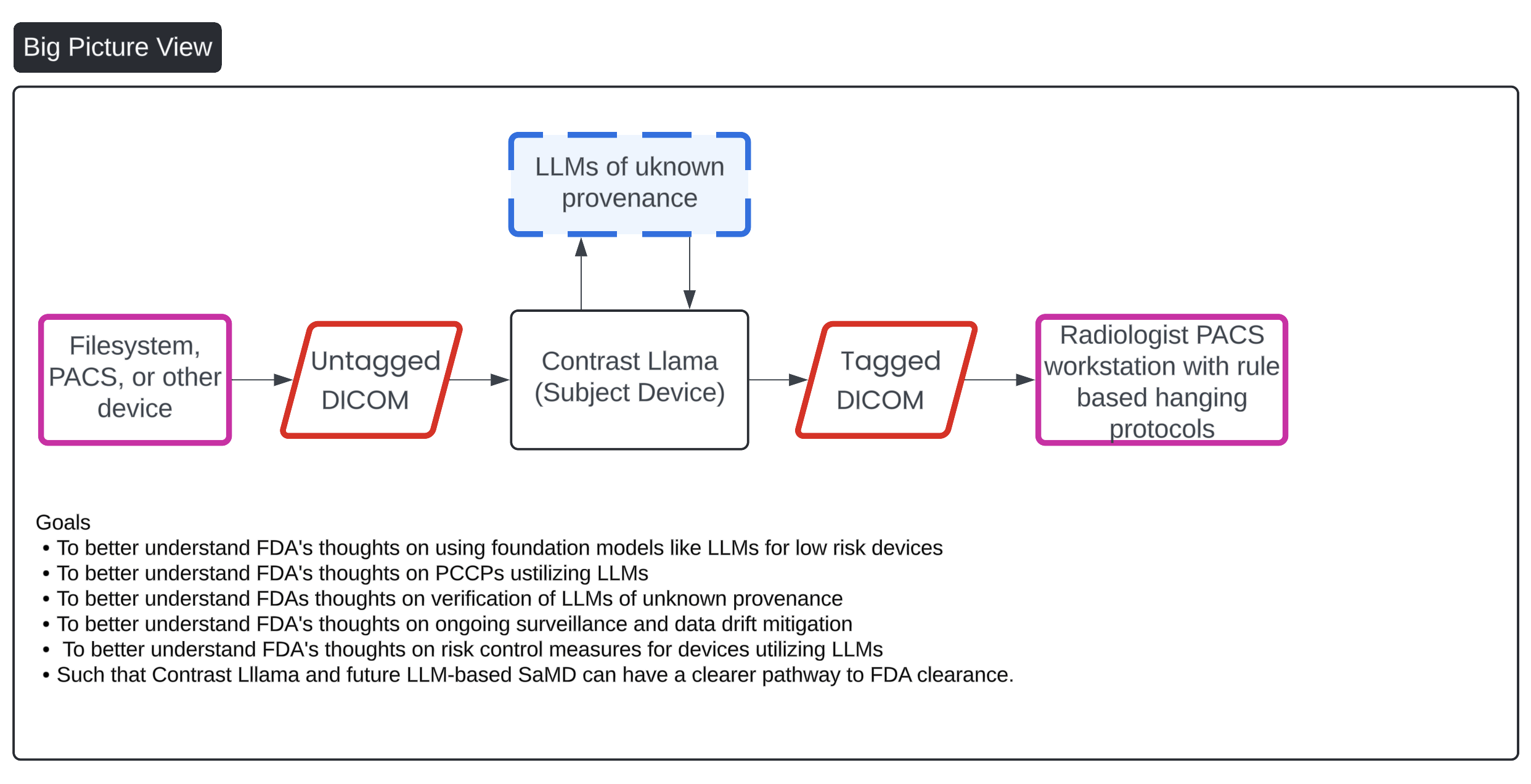
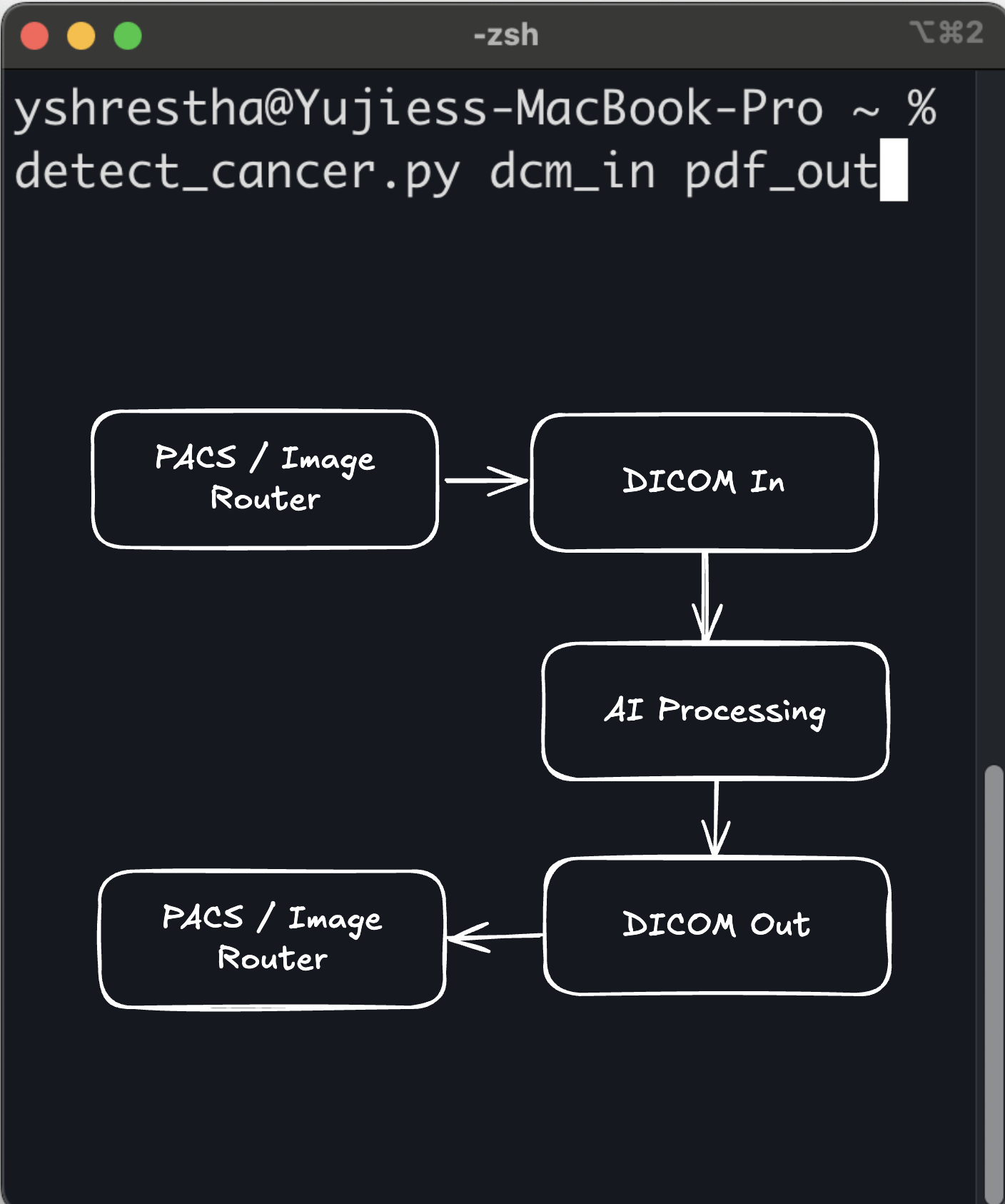
Command Line as a Medical Device (CLaMD) represents a streamlined approach to FDA clearance for medical software. By utilizing ...
Yujan Shrestha
ArticleRegulatory
To provide quality service to our clients, we need to do more than produce quality work.
J. David Giese
ArticleCybersecurity
In this article, we share a nuanced approach to avoiding sharing sensitive information in an SBOM.
J. David Giese
GuidanceRegulatory
This guidance applies to sponsors designing or submitting clinical studies for medical devices. It highlights the importance of...
Valeriia Zakrasniana
VideoRegulatory
The discussion in the transcript revolves around the FDA’s Draft Guidance on AI in Medical Devices, focusing on regulatory cons...
 Matt Hancock
Matt Hancock
VideoAI/ML
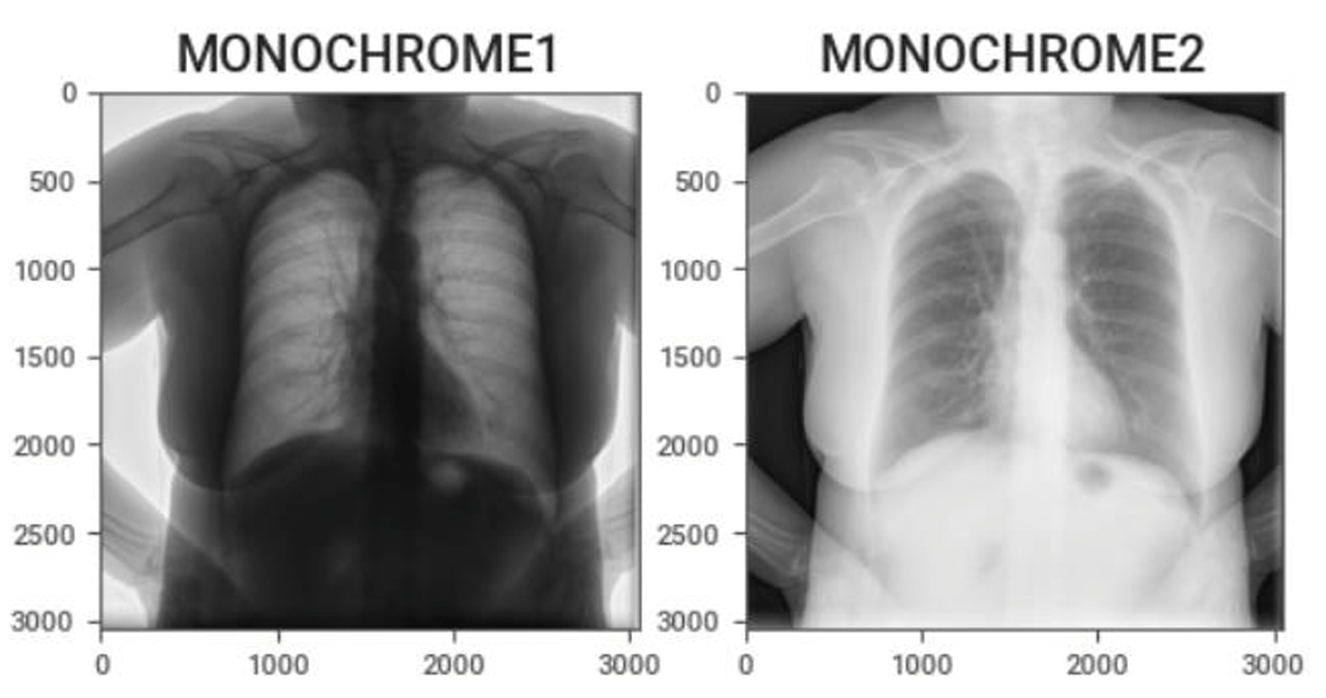
Kris Huang led a discussion on key image processing techniques in medical imaging, focusing on CT and MRI scans. Topics include...
 Kris Huang
Kris Huang
GuidanceRegulatory
Applicable to sponsors submitting PMAs for medical devices and offers a flexible, staged approach to review. By allowing earlie...
Valeriia Zakrasniana
GuidanceRegulatory
Applies to manufacturers of AI-enabled medical devices and outlines expectations for managing changes to AI software across its...
VideoRegulatory
A panel discussion about the FDA's Predetermined Change Control Plan (PCCP) guidance, particularly its application to AI-enable...
Yujan Shrestha & 2 others
VideoAI/ML
The document details the process and significance of Clinical Performance Assessments (CPAs) for medical devices, focusing on t...
 Ethan Ulrich
Ethan Ulrich
VideoAI/ML
Discussion covered key takeaways from the RSNA conference, balancing safety with speed, and setting risk thresholds. Topics inc...
Yujan Shrestha & 2 others
VideoSoftware
The discussion outlines the evolution of FDA regulations, common violations, and the enforcement structure. It emphasizes the i...
 George Hattub
George Hattub
VideoAI/ML
This is a recording of the talk Yujan gave at the RSNA AI Theater
Yujan Shrestha
Video
The growing role of AI in regulatory affairs, emphasizing its potential to enhance efficiency while requiring careful oversight...
Yujan Shrestha
Video
Yujan discusses pragmatic postmarket surveillance to FDA advisory committee on GenAI
Yujan Shrestha
VideoAI/ML
The discussion explores generative AI (GenAI) in medical devices, focusing on FDA's considerations for regulation, risks, and t...
Yujan Shrestha & J. David Giese
VideoCybersecurity
The conversation between Yujan Shrestha and J. David Giese focuses on making threat modeling more approachable and less intimid...
Yujan Shrestha & J. David Giese
VideoAI/ML
Dr. Elena Sizikova's presentation highlights the role of synthetic data in addressing data scarcity and privacy challenges in m...
Yujan Shrestha
VideoCybersecurity
We discuss challenges in medical device cybersecurity, including balancing FDA guidelines with practical hospital and manufactu...
Yujan Shrestha & J. David Giese
VideoAI/ML
Yujan and David discussed the topic of FDA's performance in regulating AI medical devices, focusing on the ideal regulatory sit...
Yujan Shrestha & J. David Giese
Article
Explores premarket evaluation, risk management, and postmarket monitoring of AI virtual scribes in healthcare, addressing new c...
Yujan Shrestha
VideoAI/ML
A 10x talk exploring the basics of prompt engineering, followed by an open-ended discussion.
VideoAI/ML
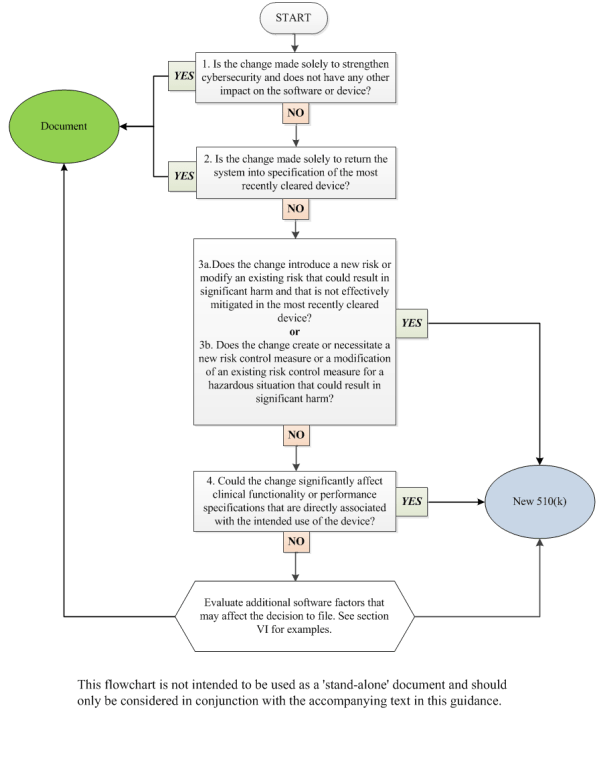
The talk covers device changes and their regulatory implications using FDA guidance. It focuses on two key documents for decidi...
 Mary Vater
Mary Vater
ArticleDICOM
A practical guide for integrating software with VistA, the VA’s PACS. This guide is based on our experience working through the...
ArticleRegulatory
This checklist is meant to assist in the review of a 510(k) submission. It is a version of the checklist we use internally at I...
Meri Martinez & 2 others
ArticleAI/ML
The article outlines a method for documenting AI/ML algorithms for FDA pre-submissions. It emphasizes clear algorithm descripti...
Yujan Shrestha
ArticleCybersecurity
A survey that should give you a feeling as to whether you’re cybersecurity documentation is ready for an FDA marketing submission.
J. David Giese
GuidanceRegulatory
Provides detailed recommendations on implementing Predetermined Change Control Plans (PCCPs), which allow manufacturers to make...
ArticleCybersecurity
How do you avoid slowing down your FDA marketing submission (510(k), De Novo, or PMA) with cybersecurity problems? This article...
Yujan Shrestha & George Hattub
ArticleSoftware
Overview of SMART on FHIR and when it’s a good approach to integrating your software into the clinical workflow. It includes a ...
ArticleAI/ML
Tips and FAQs on how to get a foundation model FDA 510(k) or De Novo cleared.
Yujan Shrestha
ArticleCybersecurity
A technical exploration and discussion of DICOM Part 15 Security
ArticleCybersecurity
How safe is DICOM in the wild? As you develop your medical imaging device or service, prepare for the standards and regulations...
ArticleRegulatory
How to Write a Device Description for a 510(k)
ArticleRegulatory
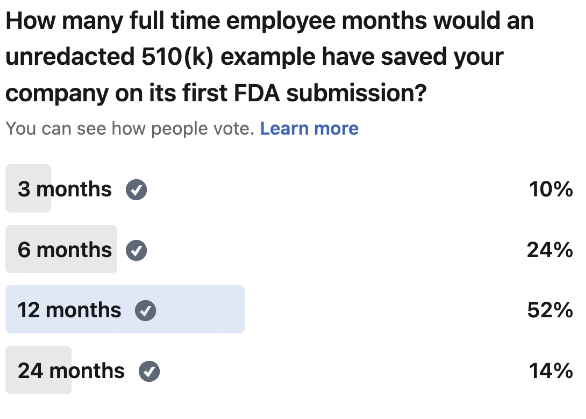
A comprehensive guide to SaMD 510(k) submissions, featuring an unredacted 510(k) as a practical example to illustrate FDA submi...
Yujan Shrestha
ArticleRegulatory
An in-depth guide to navigating the use of off-the-shelf software (OTS) in the medical device industry. OTS software is general...
J. David Giese
ArticleRegulatory
Creating a shared glossary is easier said then done. In this article, I explore common pitfalls and propose solutions to creati...
J. David Giese
ArticleCybersecurity
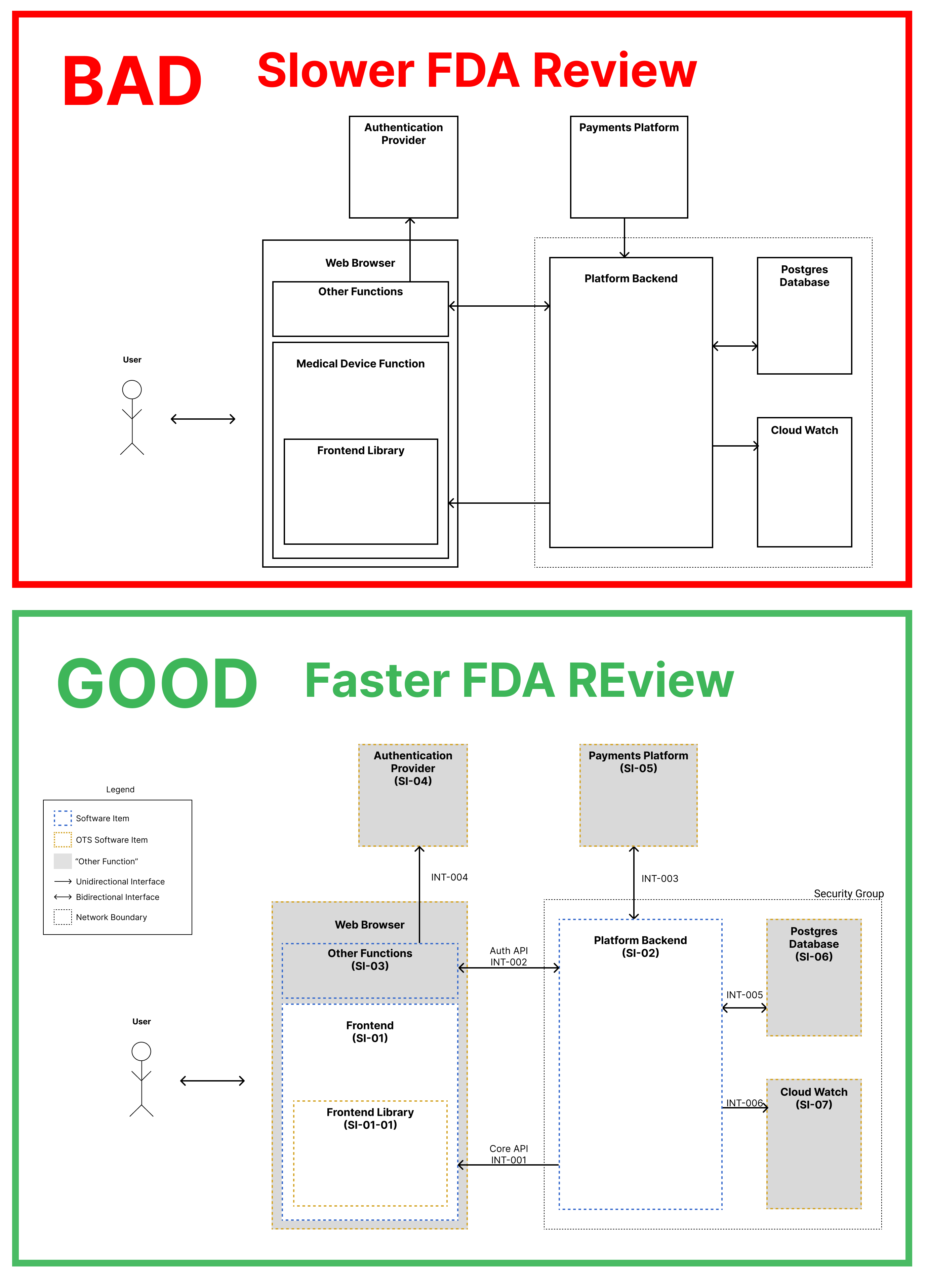
A short article that attempts to explain why badly made diagrams will confuse FDA reviewers and slow down your submissions. Inc...
J. David Giese
ArticleRegulatory
Need to write a user manual for a medical device? Not sure where to start? This article can give you a place to start
Yujan Shrestha
ArticleAI/ML
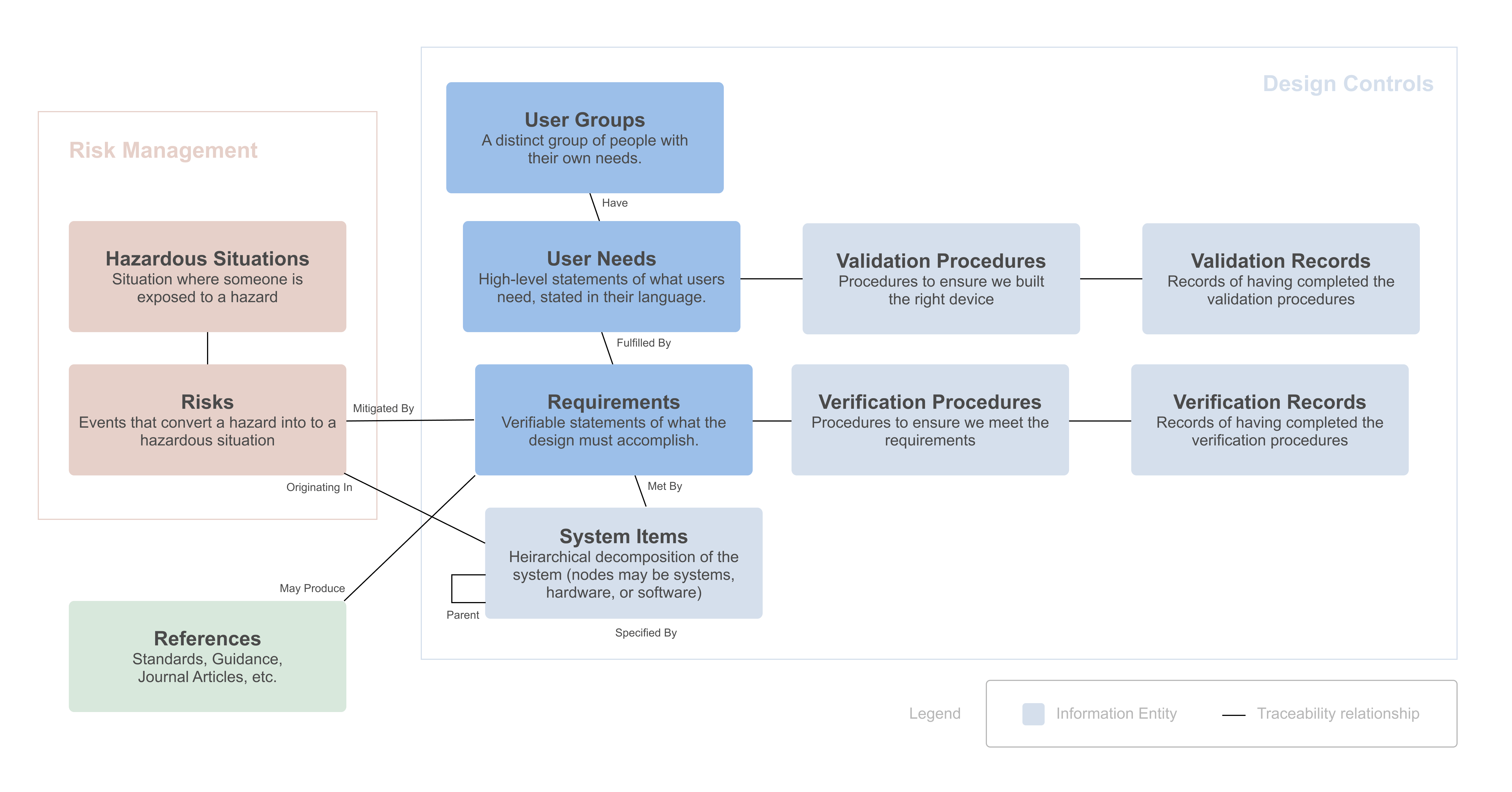
What is the difference between user needs and requirements?
Yujan Shrestha
ArticleNews
April Fools 2023 Joke: New FDA AI/ML Draft Guidance: Recommendations and Risk Assessment of Generative (AI/ML)-Enabled Quality ...
J. David Giese
ArticleAI/ML
Learn how to avoid invalidating your test dataset while still allowing yourself some flexibility to re-use the test data.
J. David Giese
ArticleRegulatory
This article discusses engineering and regulatory factors to consider when deciding to rewrite or reuse existing prototype code...
J. David Giese
VideoAI/ML
This video discusses the potential avenues for getting generative AI applications cleared through FDA.
Yujan Shrestha
ArticleAI/ML
This article outlines the process of developing an AI/ML algorithm from scratch and getting it FDA cleared. It covers the four ...
Yujan Shrestha
ArticleRegulatory
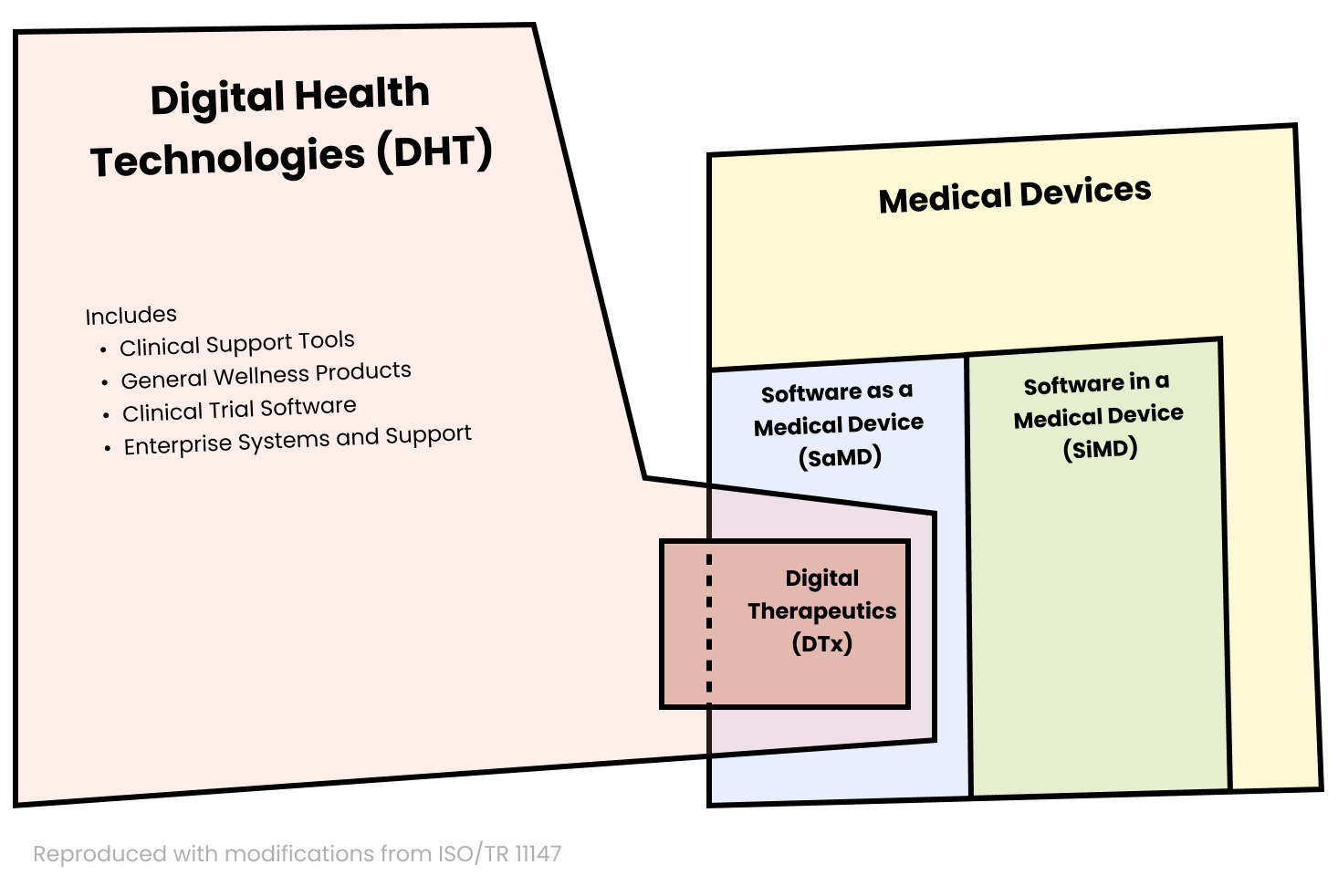
An overview of best practices and regulatory strategies related to digital therapeutics.
J. David Giese
ArticleRegulatory
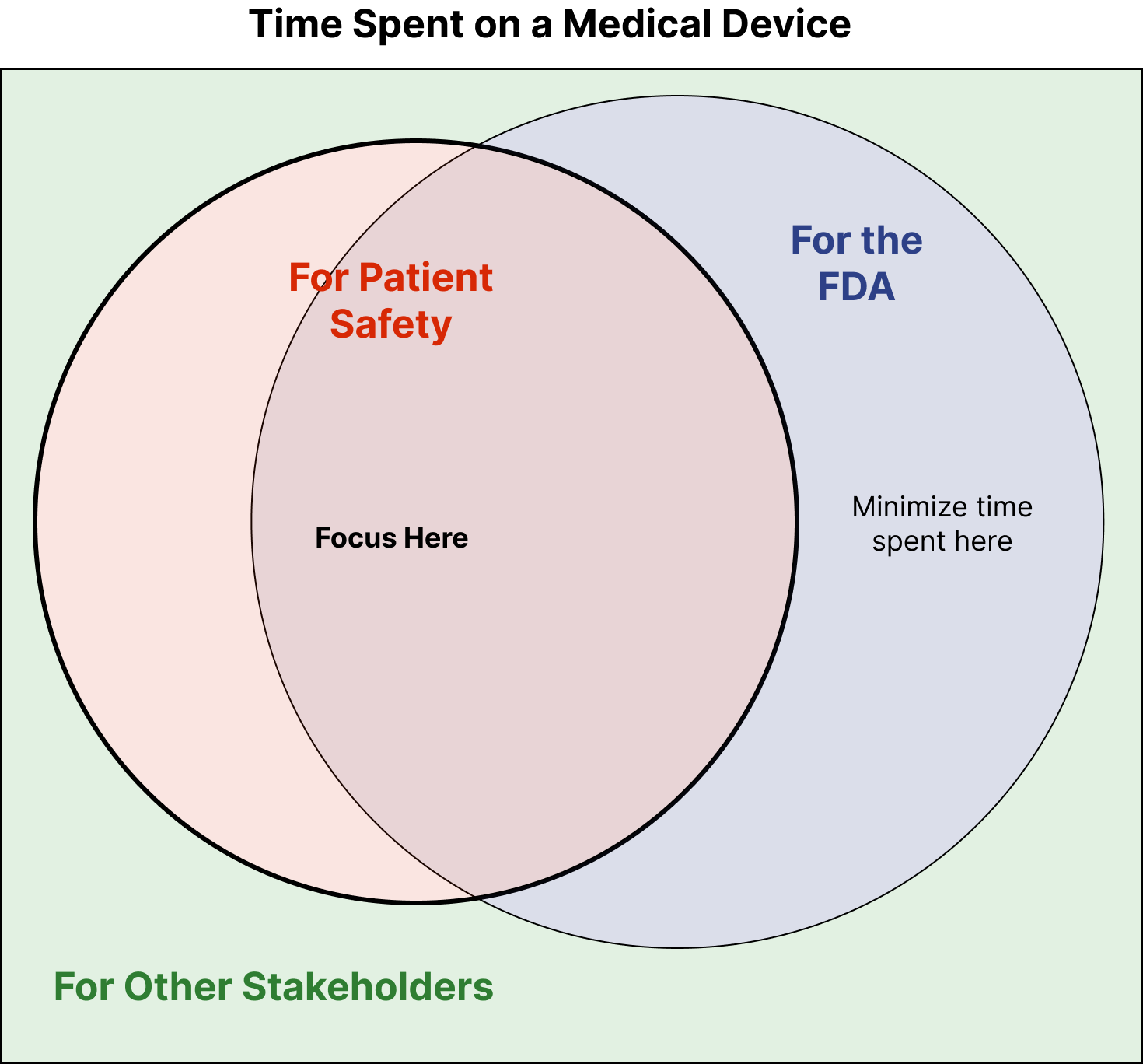
If you’ve ever been working on a medical-device and have thought, “this is a waste of time,” this article is for you. It will p...
J. David Giese
ArticleRegulatory
This article provides an overview of FDA’s webinar regarding a set of guidance documents that were recently released by FDA as ...
Meri Martinez
ArticleCybersecurity
This is a redline between the 2022 Draft cybersecurity guidance and the final 2023 cybersecurity guidance. It should be useful ...
J. David Giese & Meri Martinez
ArticleRegulatory
Medical-device software release frequency is a common question, particularly from software engineers familiar with agile develo...
J. David Giese & Jim Luker
GuidanceRegulatory
This guidance document explains how medical device manufacturers can incorporate off-the-shelf software (OTS) into their device...
ArticleAI/ML
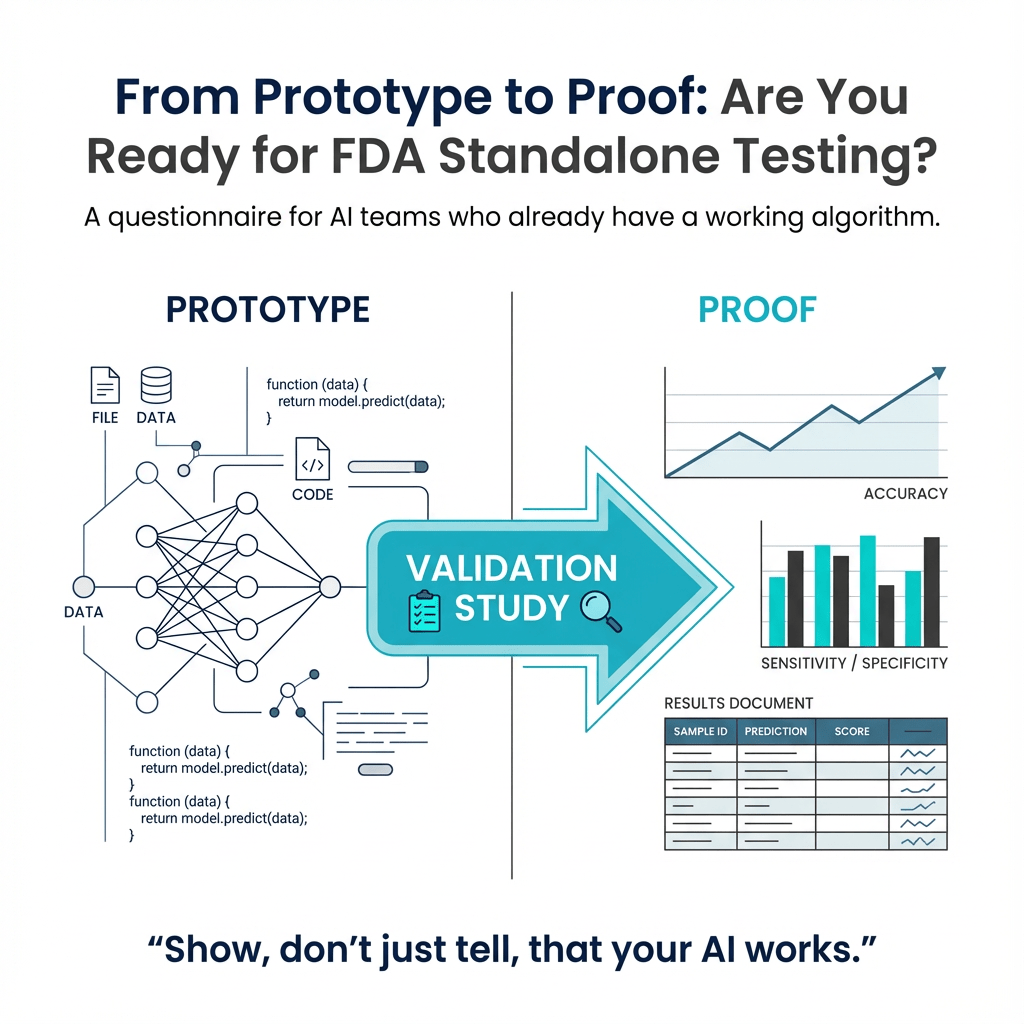
This article provides a questionnaire for those looking to get their AI algorithm FDA cleared. It covers topics such as validat...
Yujan Shrestha
ArticleSoftware
This article explains why regulatory consultants who understand software can save your engineers time and help get your medical...
J. David Giese
ArticleAI/ML
This article outlines the challenges of bringing radiology AI/ML to market, from clinical workflow to IT installation. It provi...
J. David Giese
GuidanceRegulatory
Describes all of the documents you need to include in 510(k) submissions for medical devices containing software functions. It’...
ArticleRegulatory
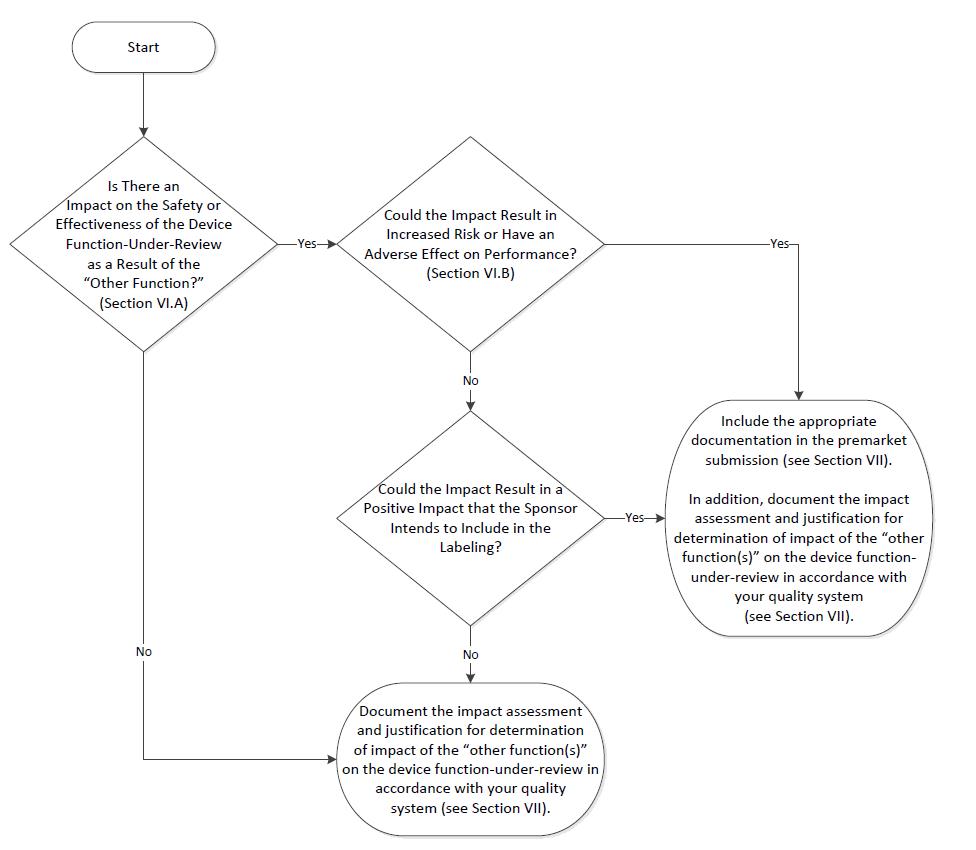
A distillation of the 27 page FDA guidance document ‘Multiple Function Device Products: Policy and Considerations’.
GuidanceRegulatory
This guidance discusses the FDA’s Q-Submission program, which can be used to get feedback on your predicate device selection, p...
VideoAI/ML
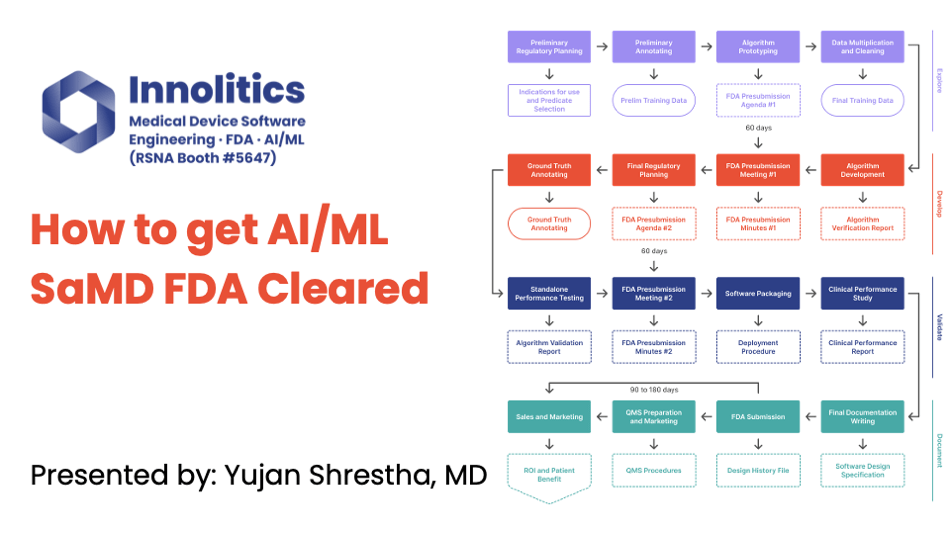
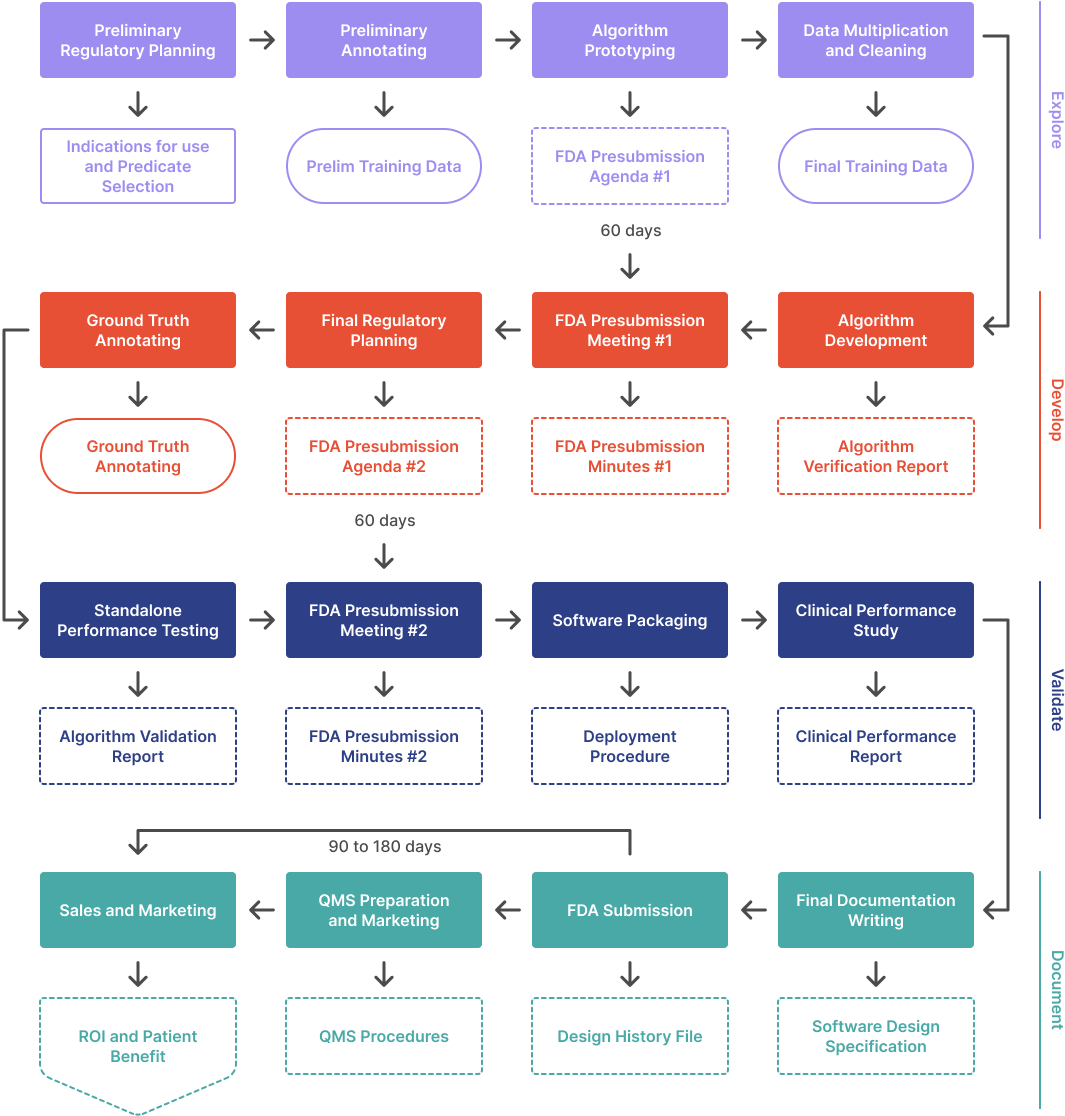
This webinar covers the four phases of the regulatory process: Explore, Develop, Validate, and Document. It also discusses the ...
Yujan Shrestha
VideoSoftware
Learn from a real-world Medtech OS software bug and explore various debugging techniques that can help you navigate through sof...
J. David Giese
VideoRegulatory
We talk through a strategy for speeding up 510(k) submissions by using a letter to file for certain features that wouldn't trig...
J. David Giese
VideoRegulatory
We talk through a strategy for speeding up 510(k) submissions by using a letter to file for certain features that wouldn't trig...
J. David Giese
VideoRegulatory
What is traceability? How does Notion help you handle it? How does Notion's flexibility empower medical device startups in ways...
J. David Giese
VideoRegulatory
A quick introduction to writing user needs and requirements for a medical device.
J. David Giese
VideoSoftware
This video explains why code reviews are essential for growing a 10x software engineering team and how to do code reviews well.
J. David Giese
GuidanceRegulatory
This FDA guidance document provides advice for making deficiency requests for medical device marketing applications in accordan...
GuidanceAI/ML
This FDA guidance applies to AI/ML-enabled radiology software that assists in the detection of disease. Many of the principles ...
GuidanceAI/ML
This guidance is describes FDA’s thoughts on evaluating the performance of CADe radiological devices. Designing standalone and ...
GuidanceRegulatory
This FDA guidance document outlines the policy for medical device data systems, medical image storage devices, and medical imag...
GuidanceAI/ML
This document outlines the technical performance assessment required for premarket submissions of quantitative imaging devices ...
GuidanceAI/ML
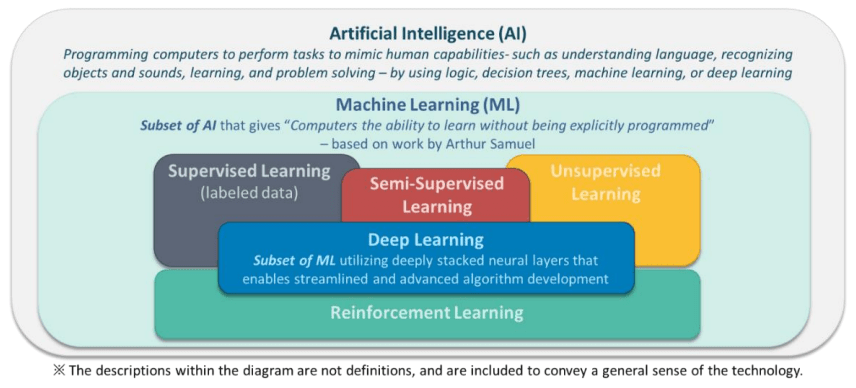
Introduces standardized terms and definitions to foster a consistent understanding across the industry for ML-enabled devices.
GuidanceRegulatory
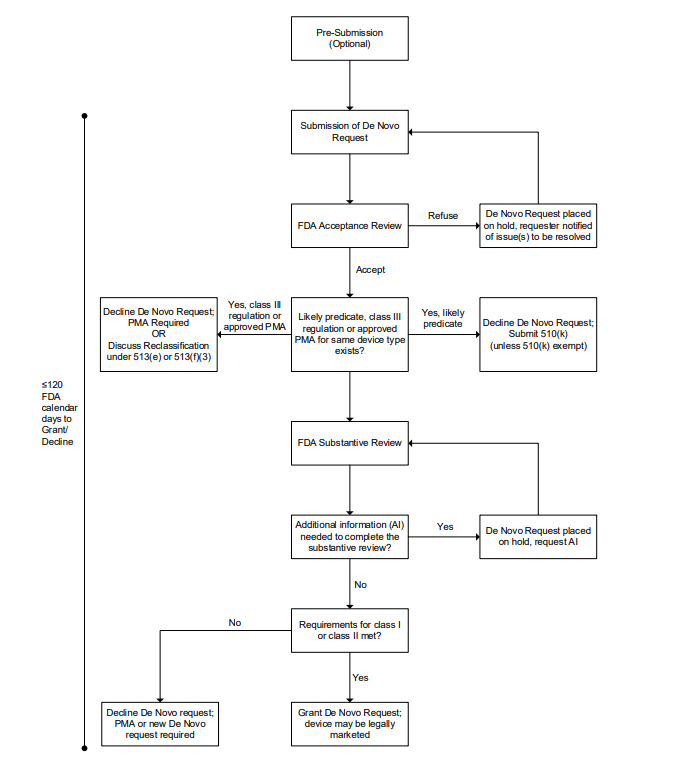
Explains how to request classification into Class I or II through the De Novo pathway, providing a streamlined alternative to t...
VideoCybersecurity
Security vulnerabilities in medical devices can have devestating consequences. They can lead to patient injuries and in the wor...
ArticleAI/ML
This article outlines Innolitics' process for developing machine learning algorithms for FDA-cleared medical devices. It covers...
Yujan Shrestha &
GuidanceRegulatory
This guidance recommends approaches that sponsors of clinical trials intended to support a new drug application or a biologics ...
GuidanceRegulatory
This FDA guidance explains the regulatory approach and policy for multiple function device products, which are products that co...
GuidanceRegulatory
This guidance applies to in vitro companion diagnostic (CDx) devices intended for use with oncology drugs. It is relevant when ...
Valeriia Zakrasniana
GuidanceRegulatory
Outlines FDA’s criteria for accepting and filing a PMA application. The guidance should help avoid delays, ensures efficient co...
Valeriia Zakrasniana
GuidanceRegulatory
This guidance provides crucial information for developers and manufacturers on how the FDA classifies and regulates software fu...
GuidanceRegulatory
When you’re developing a product that may be a medical device, one of the first questions to ask is: Is it a medical device? Us...
GuidanceRegulatory
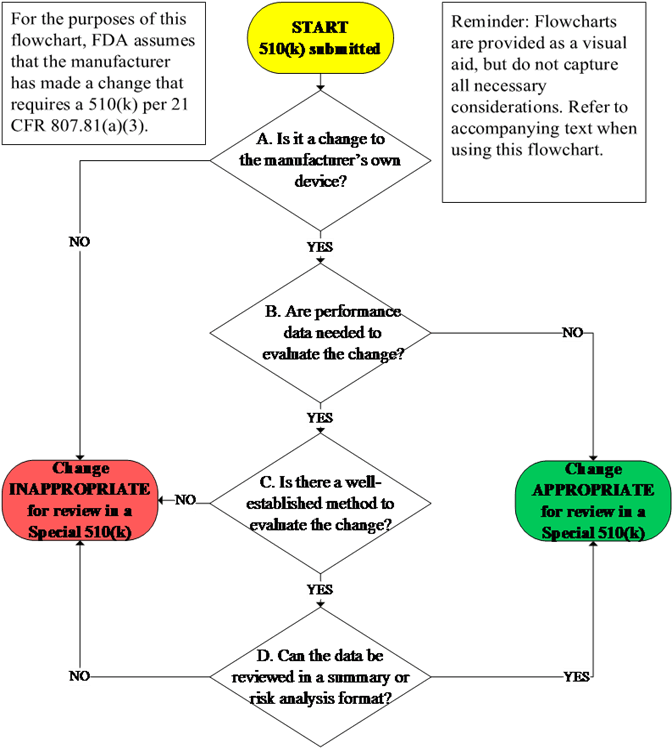
The Special 510(k) is an optional pathway for certain well-defined device modifications where a manufacturer modifies its own l...
GuidanceRegulatory
This guidance describes the abbreviated 510(k) program, which may be chosen when the device can be validated with FDA guidance ...
GuidanceRegulatory
This FDA guidance document provides a general framework for formatting and content of Traditional or Abbreviated 510(k) submiss...
GuidanceRegulatory
Applies to entities that service or modify medical devices and helps determine when those activities qualify as “remanufacturin...
Valeriia Zakrasniana
GuidanceRegulatory
The FDA guidance on the least burdensome approach—the minimum amount of information necessary to adequately address a relevant ...
GuidanceRegulatory
This FDA guidance explains the relationship between standards and regulations and how to properly declare conformance to standa...
GuidanceRegulatory
This FDA guidance provides context regarding device labeling. Labeling, which includes advertising, is important to consider ea...
GuidanceRegulatory
This FDA guidance document describes the regulation of medical device accessories and provides guidance on how accessories are ...
Guidance
This document details the necessary processes for assessing the safety, effectiveness, and performance of SAMD, including the s...
GuidanceRegulatory
This FDA guidance document provides manufacturers of medical devices with information on when a new premarket notification (510...
GuidanceRegulatory
Applies when designing and submitting clinical studies for medical devices, especially those seeking FDA approval or clearance....
Valeriia Zakrasniana
GuidanceCybersecurity
The FDA guidance discussing risks and documentation for pre-market submissions when devices must inter-operate with other devic...
GuidanceCybersecurity
This FDA guidance outlines recommendations for managing cybersecurity vulnerabilities in medical devices, including a risk-base...
GuidanceRegulatory
This document provides guidance for the FDA staff and medical device industry on how to navigate the benefit and risk factors w...
GuidanceRegulatory
Applies to clinical trials supporting FDA applications for drugs, biologics, and devices. It standardizes how race and ethnicit...
Valeriia Zakrasniana
GuidanceRegulatory
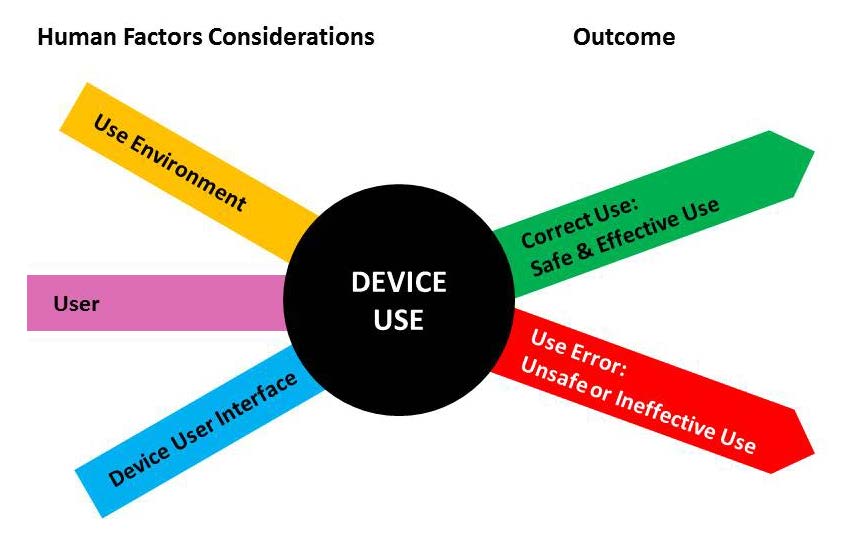
This FDA guidance document provides recommendations for medical device manufacturers to minimize potential use errors and resul...
GuidanceRegulatory
Applies to companies developing diagnostic tests that guide the safe and effective use of a corresponding therapeutic product. ...
Valeriia Zakrasniana
GuidanceRegulatory
This FDA guidance for industry and FDA staff about the current review practices for premarket notification (510(k)) submissions...
GuidanceRegulatory
This FDA guidance document describes the types of communication that occur during the review of medical device submissions. It ...
GuidanceRegulatory
The FDA guidance regarding Design Considerations for Pivotal Clinical Investigations for Medical Devices.
GuidanceRegulatory
This document provides information on how device product codes are used in various FDA program areas to regulate and track medi...
GuidanceRegulatory
Provides essential recommendations for presenting results from studies evaluating diagnostic devices. It covers qualitative dia...
GuidanceCybersecurity
This FDA guidance outlines how manufacturers should maintain the cybersecurity of medical devices that use off-the-shelf softwa...
GuidanceRegulatory
This FDA guidance outlines general validation principles that are applicable to the validation of medical device software or th...
J. David Giese
GuidanceRegulatory
This guidance serves to assist manufacturers in their development, and to assist Center reviewers in their review and evaluatio...
GuidanceRegulatory
The big old FDA Design Controls guidance from 1997, transcribed into a linkable HTML document.
J. David Giese
There are no resources matching these filter criteria.